Neural Theory
Migraine is now thought to be an episodic neurological disorder characterized by paroxysmal symptoms. According to neural theory, it is thought that migraine attacks are triggered by excessive neocortical cellular excitability. The neuronal hyperexcitability is thought to transition to CSD. Leao first demonstrated the CSD phenomenon in the 1940s.[24,25] Due to advances in neuronal excitability studies, CSD is becoming increasingly accepted as the pathophysiological basis for migraine aura and the trigger for migraine headaches. CSD can be defined as the rapid and nearly complete depolarization of a sizable population of cortical neurons with massive efflux of K+ ions from intracellular to extracellular compartments (Figure 2). CSD has several typical characteristics. The process represents a regenerative all-or-none process that spreads very slowly as a wave in the brain.[26]

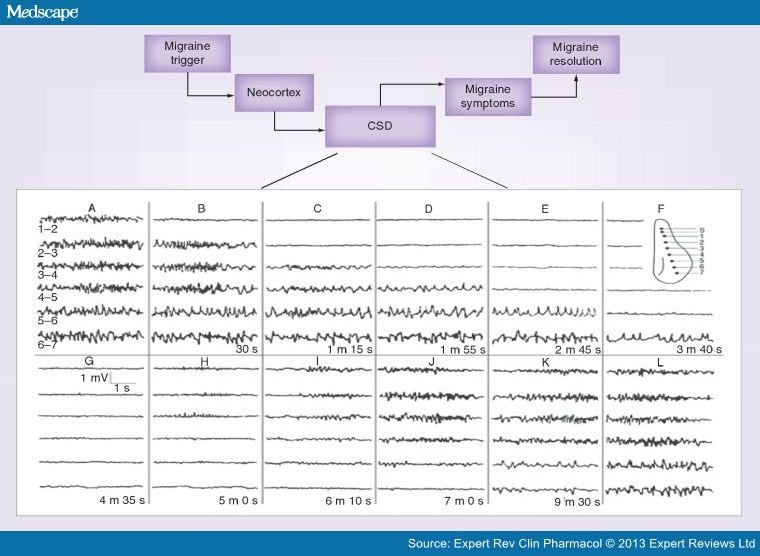
Figure 2.
Schematic illustration of the migraine pathology based on the CSD in the neocortex. CSD has been suggested as the characteristic pathophysiologic mechanism of migraine and is thought to be the neural correlate of migraine aura and may be a trigger for migraine pain. The inset shows the demonstration of the spread of CSD in the rabbit neocortex. CSD was elicited by electrical stimulation applied via an electrode at S and simultaneous recordings are illustrated in the CSD, which spreads very slowly as a wave in the cortex. Although the precise molecular basis of CSD remains unknown, the potassium theory and alternate theory are utilized to explain the phenomenon. As per potassium theory, a massive efflux of K+ creates depolarization of neurons in a slow paroxysmal fashion. There is a brief period of intense firing activity as the wave begins (caused by K+ elevations in the interstitial space that lead to the depolarization and excitation of adjacent neurons) that precedes the depression of neuronal activity (mostly due to liberation of more K+ from neurons). The alternate theory is ascribed to glutamate release, thereby leading to neuronal synchronization (excitation) that precedes depressed activity. Many antiepileptics such as valproate, topiramate and gabapentin that suppress CSD are useful for migraine prevention.CSD: Cortical spreading depression.Reproduced with permission from [24].
CSD, observed across a broad range of species from mouse to man, is a transient depression of all spontaneous and evoked activity in neural tissue, often preceded by a brief burst of action potential firing, which slowly propagates in brain tissue centrifugally from the point of origin (Figure 2).[27] The hallmarks of CSD are the complete and prolonged membrane depolarization of both neurons and glia, and the associated massive K+ efflux increasing the extracellular K+ concentration ([K+]e) from a resting level of 3–4 to >40 mM. This is accompanied by a slow negative extracellular potential shift (DC shift), which is 10–30 mV in amplitude, whereas the membrane potential and [K+]e are usually restored within a minute via Na+/K+-ATPase activation, as well as K+ clearance by astrocytes and diffusion. Complete recovery of synaptic activity may take up to 10 min in the wake of a CSD. Although the actual channels mediating the ion fluxes during CSD remain unclear, it is thought that the surge in local [K+]e depolarizes adjacent neurons and glia, and in this way the depolarization spreads. The resulting calcium influx may promote the release of glutamate, which is believed to facilitate CSD propagation. The speed of CSD propagation ranges from 2 to 5 mm/min in the cerebral cortex.[28,29] In rodents, the CSD can be elicited by cortical application of 3M KCl solution.[30] Antiepileptic drugs and anesthetics can suppress the CSD propagation.[30–35]
Expert Rev Clin Pharmacol. 2013;6(3):271-288. © 2013 Expert Reviews Ltd.