
Abstract
Purpose
Timely diagnosis and identification of etiology of pediatric mild-to-moderate sensorineural hearing loss (SNHL) are both medically and socioeconomically important. However, the exact etiologic spectrum remains uncertain. We aimed to establish a genetic etiological spectrum, including copy-number variations (CNVs) and efficient genetic testing pipeline, of this defect.
Methods
A cohort of prospectively recruited pediatric patients with mild-to-moderate nonsyndromic SNHL from 2014 through 2018 (n = 110) was established. Exome sequencing, multiplex ligation-dependent probe amplification (MLPA), and nested customized polymerase chain reaction (PCR) for exclusion of a pseudogene, STRCP, from a subset (n = 83) of the cohort, were performed. Semen analysis was also performed to determine infertility (n = 2).
Results
Genetic etiology was confirmed in nearly two-thirds (52/83 = 62.7%) of subjects, with STRC-related deafness (n = 29, 34.9%) being the most prevalent, followed by MPZL2-related deafness (n = 9, 10.8%). This strikingly high proportion of Mendelian genetic contribution was due particularly to the frequent detection of CNVs involving STRC in one-third (27/83) of our subjects. We also questioned the association of homozygous continuous gene deletion of STRC and CATSPER2 with deafness–infertility syndrome (MIM61102).
Conclusion
Approximately two-thirds of sporadic pediatric mild-to-moderate SNHL have a clear Mendelian genetic etiology, and one-third is associated with CNVs involving STRC. Based on this, we propose a new guideline for molecular diagnosis of these children.
Similar content being viewed by others

INTRODUCTION
Compared with severe-to-profound hearing loss, mild-to-moderate sensorineural hearing loss (SNHL) can easily be missed or ignored. When congenital or prelingual hearing loss is detected, with proper management it can alleviate substantial medical and educational sequelae and socioeconomic burden. However, neglected mild-to-moderate SNHL could potentially cause more significant disability in academic achievement than those with more severe but well-managed SNHL.1,2,3 The first step toward appropriate management of pediatric mild-to-moderate SNHL is establishing a timely, correct audiological and etiologic diagnosis. Prognosis prediction and plans for comprehensive rehabilitation are only possible after a clear etiologic diagnosis.
Recently, many new deafness genes have been cloned (https://hereditaryhearingloss.org/) and CNVs have been spotlighted as an important molecular cause of mild-to-moderate SNHL, especially for DFNB16, which is caused by an alteration of STRC.4,5,6 However, despite this era of precision medicine, there seems to be a lack of interest in the etiologic spectrum of pediatric mild-to-moderate SNHL, especially with respect to the sporadic form. To the best of our knowledge, limited studies have addressed the etiology of this disease, focusing on the genetic cause,7,8 and none of these studies have intensively evaluated the presence of copy-number variations (CNVs); hence, the genetic cause might have been underestimated. Furthermore, those studies also included a significant portion of adults with SNHL with a more heterogeneous etiology,5,6 precluding the revelation of a true genetic spectrum of pediatric mild-to-moderate SNHL. Therefore, a more comprehensive investigation into the molecular etiology of sporadic mild-to-moderate pediatric SNHL may be warranted, incorporating recent discoveries and technical advances.
This prospective study, incorporating the largest mild-to-moderate SNHL pediatric cohort and the most sophisticated molecular testing protocol to date, reports that a significant portion of this condition is caused by a Mendelian genetic etiology, more specifically, CNVs.
MATERIALS AND METHODS
Ethical considerations
This study was approved by the institutional review boards (IRBs) of the Seoul National University Bundang Hospital (SNUBH) (IRB-B-1007-105-402) and the Seoul National University Hospital (SNUH) (IRBY-H-0905-041-281). We obtained written informed consent from either the parents of children or the participants themselves.
Eligibility criteria and clinical evaluation
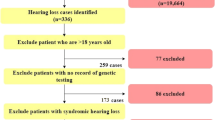
We prospectively recruited pediatric patients with bilateral symmetrical nonsyndromic SNHL at the otolaryngology clinics of two institutions from January 2014 through December 2018, only when the mean hearing threshold was 26 dB or higher, and when the onset age was clearly documented by audiological tests to be prior to age 15 years (SNU-Bi-NS-Child-Spo/AR SNHL cohort [n = 320]) (Fig. 1). Clinical characteristics (medical history, physical examination) and audiologic assessments (pure‐tone audiometry, auditory brainstem response [ABR], auditory steady state response [ASSR], and speech evaluation) were reviewed. The hearing threshold was calculated by averaging the thresholds of 0.5, 1, 2, and 4 KHz.

Among the SNU-Bi-NS-Child-Spo/AR SNHL cohort, only those with a hearing threshold of ≤55 dB and those who consented to molecular genetic testing are registered as SNU-mild-to-moderate SNHL. PT/SA Pure-tone audiometry/ Speech audiometry, ABR-T Auditory Brainstem Response Threshold, ASSR Auditory Steady State Response.
Only cases with sporadic or autosomal recessive (AR) inheritance pattern were included. Cases with apparent vertical transmission from parents to children by autosomal dominant (AD) inheritance were excluded. Additionally, syndromic SNHL, asymmetrical SNHL with 20 dB or more difference between bilateral ears, and mixed type hearing loss were excluded from this study.
The SNU-Bi-NS-Child-Spo/AR SNHL cohort was then divided into two groups, based on the degree of SNHL: one with mild-to-moderate SNHL (≤55 dB hearing threshold) (n = 110) and the other with moderately severe-to-profound SNHL (>55 dB hearing threshold) (n = 210) (Fig. 1). Only those who manifested mild-to-moderate SNHL and consented to molecular genetic testing were finally included (n = 83), providing the youngest (mean age of 7.63 years of age) and the largest genetically tested mild-to-moderate pediatric bilateral SNHL cohort to date.
DNA preparation and molecular genetic test
Whole blood (10 ml) was obtained from all probands and, if possible, their siblings and parents for the segregation study. Genomic DNA was extracted from the peripheral blood.9 Then, molecular genetic testing was performed using multiple genetic tools: multiplex ligation-dependent probe amplification (MLPA) of STRC, exome sequencing, and long-range (LR) polymerase chain reaction (PCR) of whole STRC, if necessary. The presence of a pseudogene (pSTRC), which is 99.6% identical to a true gene (STRC), has been reported to be associated with low next-generation sequencing (NGS) mapping quality.10 To compensate for the pitfalls of NGS (i.e., exome sequencing), MLPA, which could detect CNVs, and LR-PCR, which could exclude pseudogene contamination, were implemented in the diagnostic process.
Multiplex ligation-dependent probe amplification
The SALSA MLPA P461 DIS probemix kit (MRC-Holland, Amsterdam, the Netherlands) was used to detect CNVs of STRC, as suggested by the manufacturer. Amplification products were run on an ABI PRISM 3130 Genetic Analyzer (Applied Biosystems, Foster City, CA) and the results were analyzed using Gene Marker 1.91 software (SoftGenetics, State College, PA).
Exome sequencing
Exome sequencing was performed11,12,13 and the obtained reads were mapped onto the University of California–Santa Cruz (UCSC) hg19 reference genome assembly, employing Lasergene 14 software package (DNASTAR, Madison, WI, USA).
Long-range PCR/nested PCR
To prevent the primer from attaching to the STRCP sequences, two LR-PCR products were generated for subsequent nested PCR and primers were designed.14,15,16 LR-PCR was performed using the Qiagen LongRange PCR Kit (Qiagen, Hilden, Germany).16
Sanger sequencing of MPZL2
For the remaining probands whose genetic diagnosis was not made through MLPA of STRC and exome sequencing, Sanger sequencing of MPZL2 in cases without exome sequencing was performed to detect causative variants in MPZL2. Specifically, the p.Gln74* variant of MPZL2 was screened, as the MPZL2 gene had been recently reported to be a deafness gene in late 2018, when the study was almost completed.17
Decision of genetic diagnosis status
Further bioinformatics analyses were performed to identify the causative variants.18,19 Global minor allele frequency (MAF) was checked using several databases, including the 1000 Genomes Project (1000 Genomes), Exome Aggregation Consortium (ExAC), and National Heart, Lung, and Blood Institute (NHLBI) Grand Opportunity Exome Sequencing Project (GO-ESP). These nonsynonymous single-nucleotide polymorphisms (SNPs) were compared against the Korean Reference Genome Database (KRGDB), consisting of 1722 Korean individuals (3444 alleles) (http://152.99.75.168/KRGDB/menuPages/firstInfo.jsp). The inheritance patterns were checked and SNPs that were incompatible with the AR pattern were excluded. To predict the pathogenicity of each variant, Sorting Intolerant from Tolerant (SIFT), PolyPhen-2, MutationTaster, Combined Annotation Dependent Depletion (CADD), and Rare Exome Variant Ensemble Learner (REVEL) analyses were performed (Table S1). The evolutionary conservation of the amino acid sequence was estimated using the GERP++ score in the UCSC Genome Browser (http://genome.ucsc.edu/). The variants that were predicted as benign by in silico prediction were excluded. The remaining SNPs were validated in other family members by Sanger sequencing for segregation study. Pathogenic variants are described according to the American College of Medical Genetics and Genomics (ACMG) 2015 guidelines and newly specified ACMG/Association for Molecular Pathology (AMP) hearing loss rules.20,21,22 “Genetically diagnosed” was judged only when two likely pathogenic or pathogenic variants were identified.
Genotyping of short tandem repeat (STR) markers
Six p.Gln74* (MPZL2) allele-linked STR markers (D11S1986, D11S1885, D11S4127, D11S4171, D11S925, and D11S4107) were chosen17 and genotyped in 81 normal-hearing controls, and 9 p.Gln74* allele carriers in the hearing-impaired cohorts to identify haplotypes, to investigate whether the mutant allele (p.Gln74*) might be a mutational hotspot or a founder allele.23
Integration of deep learning algorithms
To identify the algorithm in differentiating the auditory phenotype of DFNB16 from that of DFNB111, deep learning algorithms were developed. First, the hearing level was measured at seven frequencies (250 Hz, 500 Hz, 1 KHz, 2 KHz, 3 KHz, 4 KHz, and 8 KHz); then, hearing level was measured at six additional frequencies that are intermediates of the initial seven frequencies (in between the abovementioned frequencies). Then, a modified repeated fourfold cross-validation (CV) was performed to minimize the limitation of having a small cohort in this study and to minimize potential bias resulting from random sampling (Fig. S1).24 Next, we designed three distinct core models: (1) a basic neural network consisting of one hidden layer and one fully connected layer (FCN); (2) a long–short term memory (LSTM), which is part of the recurrent neural network (RNN) architecture; and (3) a one-dimensional convolution neural network (1D CNN). Based on these three core models, we modified several parameters and placed additional layers to make ten models.25,26
Statistical analysis
Mann–Whitney test was conducted to compare the audiometric results, depending on the genotypes. Fisher’s exact test was performed to compare the haplotype patterns around p.Gln74* (MPZL2) between the subjects with p.Gln74* and controls. All statistical tests were two-sided, and P values < 0.05 were considered statistically significant.
RESULTS
Demographic and audiologic data of SNU-mild-to-moderate SNHL
Mean age, mean hearing threshold, and proportion of male subjects in our study cohort were 7.63 years, 45.1 dB, and 51.8% (43/83), respectively.
Genotypic spectrum of SNU-mild-to-moderate SNHL
Genetic diagnosis was made in 52 subjects (52/83 = 62.7%); STRC-related deafness (DFNB16, n = 29, 29/83 = 34.9%) was the most prevalent, followed by MPZL2-related deafness (DFNB111, n = 9, 9/83 = 10.8%) (Tables 1 and 2). Four compound heterozygotes of GJB2, two homozygous SLC26A4 variants, two de novo TECTA variants, and two compound heterozygote USH2A variants were also found to be causes of deafness in our cohort. Compound heterozygotes of OTOG, PDZD7, and LOXHD1 accounted for deafness in the remaining three subjects (Table 2). p.Gln1353* and p.Leu1409Serfs25* were the most commonly detected point mutations of STRC (Table 1).
The significant contribution of STRC CNVs to DFNB16 was noted. Among the 29 DFNB16 subjects, 27 subjects carried at least one copy of CNV encompassing STRC (CNV-STRC); of these, 10 subjects carried only one copy and 17 subjects carried two copies. Exome sequencing alone without incorporation of MLPA showed a significant diagnostic discrepancy compared with combined MLPA+ exome sequencing strategy in 88.9% (16/18) of cases. With cases identified by exome sequencing alone, LR/nested PCR was essential for the confirmative genetic diagnosis to completely exclude the contamination effect from pseudogene.
For the remaining cases where a genetic etiology was not confirmed after MLPA and/or exome sequencing, screening of MPZL2 identified nine MPZL2-related deafness cases, and p.Gln74* was the most common causative variant involved in every DFNB111 case in our cohort (Table 2). Detailed information of each causative variant is described in Table S1.
Genotyping of STR markers in MPZL2
p.Gln74*-linked haplotypes of six STR markers were analyzed among the controls and hearing-impaired patients (Table S2). Variants arose from diverse haplotypes, even in patients with homozygous p.Gln74* alleles, and no haplotype was significantly associated with p.Gln74*, even when considering four (D11S1986, D11S4127, D11S925, and D11S4107) or two STR markers (D11S1986 and D11S4171) (subjects [2/9] vs. control [10/81], P = 0.439; subjects [2/9] vs. control [9/81], P = 0.302, respectively, by Fisher’s exact test) (Fig. S2 and Table S2). This argues against the presence of a single common founder allele linked to p.Gln74*. However, considering the significantly higher minor allele frequency of p.Gln74* in Koreans and Japanese compared with other populations (Table S3), our results suggest that the p.Gln74* allele could be a very old founder allele exclusively in Koreans that went through numerous recombinations across multiple generations and finally came to retain only a very narrow common haplotype that we could not detect.
Proportion of potential deafness–infertility syndrome (DIS) candidates among our DFNB16 cohorts and their semen analyses
Of the 29 DFNB16 subjects, 13 (44.8%) were diagnosed with contiguous STRC-CATSPERS2 homozygous deletion, and therefore, were potential candidates for DIS if the proband was male.27 Semen obtained from two adult subjects (SH157-341 and SB291-584), whose SNHL was detected a decade ago with continuous gene deletion of STRC and CATSPER2, was analyzed in terms of sperm motility, viability, and morphology. Both subjects had normal range of sperm counts (concentration), motility, and sperm morphology according to the criteria by the World Health Organization (WHO) (Table S4),28 against previous reports.27,29
Different audiologic phenotypes between DFNB16 and DFNB111
Descriptive statistics are summarized in Fig. 2a, b. Focus was given to DFNB16 and DFNB111, as these are the two major genotypes in our analysis. To adjust for confounding effects of age, the last audiograms for DFNB16 during the follow-up period were compared with the first audiograms for DFNB111, yielding no significant difference in age at audiogram (Fig. 2c). Specifically, there was a significant difference in hearing thresholds at 4000 Hz between the groups, showing a slightly worse hearing for DFNB111 (45.4 ± 8.8 vs. 52. 8 ± 6.3 dB, respectively for DFNB16 and DFNB111 with p value of 0.037 by Mann–Whitney test). Audiogram configurations of DFNB111 demonstrated a downsloping pattern compared with those of DFNB16, which mostly showed a flat configuration that was stable over time (Fig. 2c). Subjects with other genetic etiologies or idiopathic SNHL seemed to be associated with worse hearing thresholds than DFNB16, even with younger age at the time of audiologic test (statistically significant at 1000 Hz between DFNB16 and other genetic etiology [p = 0.03]; statistically significant at 500 and 1000 Hz between DFNB16 and idiopathic etiology [p = 0.05, 0.01 respectively] by Mann–Whitney test) (Fig. 2a, b).

(a) Demographic and audiologic data are shown according to the genotypes and it is notable that the auditory threshold at 4 kHz is statistically different between DFNB16 and DFNB111. (b) Configuration of audiograms seem to be different depending on genotype. (c) It is shown that DFNB16 and DFNB111 manifest different audiogram configuration and significantly different hearing threshold at 4 KHz measured by pure-tone audiometry.
Integration of a deep learning algorithm in the genetic diagnosis of mild-to-moderate SNHL
We observed that there was a statistically significant threshold difference in terms of audiological phenotype between DFNB16 and DFNB111, especially at 4 kHz. Given that our first-line screening aims to detect CNV-STRC (DFNB16) or MPZL2 p.Gln74* (DFNB111), we questioned whether a deep learning algorithm could help prioritize one over the other.
The result of the prediction algorithm according to deep learning models is shown in Table S5. Of the ten models, the LSTM with 64 hidden layers (LSTM 64) achieved the best average prediction accuracy (89.4 ± 4.1%) with respect to the predictability of DFNB16 as DFNB16 and DFNB111 as DFNB111.
DISCUSSION
Few studies are currently available regarding the entire etiologic spectrum, including the genetic etiologic spectrum, of pediatric mild-to-moderate SNHL. A much milder phenotype, despite its huge potential sequelae, has failed to attract interest from either otorhinolaryngologists or pediatricians. Given this, our prospective study included an invaluable cohort of SNU-mild-to-moderate SNHL, which was rigorously characterized both audiologically and genetically, serving as the youngest and largest genetic cohort exclusively devoted to pediatric mild-to-moderate SNHL to date. This study revealed that a high proportion of this sensory defect—as high as two-thirds (62.7%) of the study population—was attributable to Mendelian genetic etiology, despite very stringent and conservative bioinformatics criteria. Our prospectively obtained SNU-mild-to-moderate SNHL cohort (n = 83 families) comprised 64 simplex cases and 19 multiplex cases, minimizing selection bias toward Mendelian genetic cases. In such cases, we also elucidated that STRC and MPZL2 may play major causative roles. The SNU-mild-to-moderate SNHL cohort also identified two subjects (SB380-752 and AJ13-32) with cytomegalovirus (CMV)-related SNHL as documented by internal auditory canal magnetic resonance image (IAC-MRI) and two enlarged vestibular aqueduct (EVA) subjects with SLC26A4 variants (SB478-924 and SB479-925), suggesting a comprehensive nature of our cohort and also that the proportion of the etiolgies such as CMV infection or EVA initially presenting with mild-to-moderate SNHL but being likely to worsen later to a severe degree is non-negligible.30,31
This genetic diagnostic rate has significantly increased since our previous report,7 although we focused on milder hearing loss in this prospective study by excluding severely moderate SNHL (>55 dB). Our previous study, which used only exome sequencing but not MLPA, showed a genetic diagnosis rate of 45.4% in sporadic mild-to-moderately severe SNHL subjects;7 at this point in time, MPZL2 had not yet been reported as a deafness gene. Since then, we have refined the genetic testing pipeline to include MLPA for CNVs of STRC and LR/nested PCR to avoid pseudogene (STRCP) contamination (Fig. 3). The most common genetic cause, DFNB16, which accounts for 34.9% of all pediatric mild-to-moderate SNHL, turned out to be associated with at least one copy of CNV-STRC in 93.1% (27/29) of cases, making a detection step of CNV-STRC crucial. This is not surprising considering a high normal carrier rate of CNV-STRC among Koreans (4 of 100 alleles in in-house [SNUBH] DB) and Japanese.6 No progressive nature of hearing loss and better thresholds of DFNB16 compared with other genetic or idiopathic etiology account for the higher proportion of CNV-STRC in the SNU-mild-to-moderate SNHL cohort than previous Japanese and Czech studies, which included both adults and children.5,6 Our cohort, characterized by a much younger age at recruitment with milder hearing loss—with the exclusion of severely moderate SNHL—was likely to be preferentially composed of DFNB16. Therefore, exome sequencing should neither be the first nor the only step, especially in the molecular diagnosis of mild-to-moderate SNHL in younger ages. For example, in our previous study, one DFNB16 pedigree (SB38-75) was reported to be homozygous for the stopgain variant (c.4057C>T: p.Gln1353*, NM_153700) through exome sequencing.18 However, in our current study, MLPA of STRC revealed one copy of CNV-STRC in a trans configuration to the single heterozygous p.Gln1353* (c.4057C>T) in this family, which subsequently corrected the genotype underlying this DFNB16 family, further emphasizing the essential role of MLPA in the diagnosis of DFNB16.

The detailed process of genetic diagnosis of our cohort is described according to the proposed guideline. CNV copy-number variation, LR PCR long-range polymerase chain reaction.
The high normal carrier rate of contiguous STRC-CATSPER2 deletion or CATSPER2 deletion from our in-house DB (2/100 alleles = 2% or 3/100 alleles = 3%, respectively), as detected by MLPA, prompted us to check whether pediatric mild-to-moderate SNHL subjects with two copies of CNV-STRC-CATSPER2 are common, because this contiguous gene deletion syndrome has previously been known to cause DIS.27,32 Indeed, 13 of 83 subjects in our cohort were classified as such (Table 1). However, both probands (SH157-341, SB291-584) from adults who agreed to semen analysis showed normal sperm characteristics according to the 2010 WHO criteria (normal form sperm of 4% or higher). Moreover, in the case of SH157-341, his wife had succeeded in conceiving within one year after marriage during the study period. In 2007, when this contiguous gene deletion syndrome was first named,27,32 the lower limit of the normal range for the normal form sperm (%) was 15%. Based on this criteria, SH157-341 might have been diagnosed as DIS. However, the reference values for human semen, according to the WHO, have been steadily extended since 1980 until the last update in 2010, reflecting significant advances in in vitro fertilization (IVF) and the current description of the semen characteristics of recent fathers.28 Given this, and considering the relatively high incidence of CATSPER2 deletion in normal controls, DIS might need to be reconsidered, or renamed, to minimize unnecessary anxiety of infertility in young male adults with DFNB16 associated with STRC-CATSPER2 deletion.33 However, a firm conclusion cannot be drawn from this study; further discourse is required.
The sophisticated design of LR/nested PCR, which exclusively amplified the true gene, allowed us to circumvent another issue regarding DFNB16: pseudogene contamination. For example, in a mutant 4057th residue, the T from a true gene STRC actually serves as a wild-type sequence in the pseudogene, STRCP, and the residue resides in the region with a significantly high homology with STRCP, raising the possibility of pseudogene contamination, as previously shown.10,34 The LR/nested PCR clearly verified the presence of p.Gln1353* in the true gene, without pseudogene contamination from SB38-75 in our study (Fig. S3). In 7 of 16 DFNB16 cases with at least one point variant of STRC (SB135, SB201, SB433, SH229, SB175, SB88, and SB148), the point variant resided in a region between exons 2 and 10, which is rarely covered by NGS (exome sequencing) due to a significant sequence homology with STRCP based on in-house DB Samsung Genome Institute (SGI) (unpublished data).10 LR/nested PCR was critical to accurate genetic diagnosis in such cases, as shown in SB433-840 (Fig. S3).
As shown above, MPZL2 accounted for 10.8% of the etiologies in our mild-to-moderate SNHL cohort, and it also accounted for 17.3% of the genetically diagnosed pedigrees in our cohort. Interestingly, one variant of MPZL2, p.Gln74*, which had previously been reported,17 was found to be associated with all nine DFNB111 cases in our cohort, where eight cases (88.9%) were homozygous for this variant. Genotyping of STR markers identified this variant as a potential mutational hotspot from diverse haplotypes, which is expected to elevate the global effect of the variant in the field of deafness. However, an exclusively higher MAF of this variant in the 1000 Genomes East Asian group (0.0069) and in the Korean population (0.008) compared with other populations could raise an alternative possibility, i.e., that this is a very old founder allele in Korea that lost stretches of common haplotype among probands due to recombinations over many generations. Regardless, our results explain the significant contribution of this variant to genetic hearing loss in these ethnic groups. To the best of our knowledge, this is the first paper to report the significant genetic load of MPZL2 in a large cohort of mild-to-moderate hearing loss.
Interestingly, the p.Val37Ile variant of GJB2 was detected only in two subjects (SB261-514 and SB381-738) in this study. Presumably, the homozygous p.Val37Ile variant genotype, which is relatively rare, could cause mild hearing loss, but compound heterozygotes with p.Val37Ile on one allele and another GJB2 variant in a trans allele could be related to more severe, progressive hearing loss,8 which might not meet the inclusion criteria of our study (≤55 dB). This DFNB1 genotype would have been much more prevalent if the cohort had included subjects with severely moderate hearing loss.
The undiagnosed pediatric mild-to-moderate SNHL cases even after this rigorous molecular genetic testing could largely be attributed to either infectious causes, such as nonsyndromic CMV infection, or novel deafness genes that have not yet been identified. In our study, virtually all probands were ascertained after the age of 3 weeks; therefore, a confirmatory diagnosis of congenital CMV infection could not be made. Anticipative screening for congenital CMV coupled with molecular genetic testing might significantly increase the etiologic diagnostic rate.
Based on our prevalence data, we propose a guideline for the molecular diagnosis of pediatric mild-to-moderate SNHL (Fig. 3). MLPA of STRC or Sanger sequencing of the p.Gln74* variant in MPZL2 could be used as the first screening step for genetic diagnosis. In our study, the deep learning model trained by audiologic threshold data of DFNB16 and DFNB111 shows its potential ability to help choose one of the two tests, MLPA of CNV-STRC or Sanger sequencing of p.Gln74*, based solely on audiograms, providing future application of artificial intelligence–assisted algorithms in genetic diagnosis of hereditary deafness.
Our study revealed that despite the most stringent and conservative bioinformatics tools, pediatric-onset mild-to-moderate SNHL is caused largely by Mendelian genetic etiology (2/3). Moreover, our study showed the importance of two major genes in the genetic diagnosis of our cohort: STRC and MPZL2. We also showed that CNVs of STRC accounted for one-third (27/83 = 32.5%) of our mild-to-moderate SNHL cohort, which makes MLPA of STRC an essential step in the diagnosis of mild-to-moderate SNHL. Males with contiguous STRC-CATSPER2 gene deletion, previously named DIS, should thus not be stigmatized as infertile, and DIS may need to be re-evaluated. We propose a strategy to efficiently diagnose the genetic cause, and identify the causative variants, of pediatric mild-to-moderate SNHL to provide better counseling and help reduce the under-recognized disadvantages caused by this mild, but discernible, handicap.
References
Marschark M, Shaver DM, Nagle KM, et al. Predicting the academic achievement of deaf and hard-of-hearing students from individual, household, communication, and educational factors. Except Child. 2015;81:350–369.
Schlieper A, Kisilevsky H, Mattingly S, et al. Mild conductive hearing loss and language development: a one year follow-up study. J Dev Behav Pediatr. 1985;6:65–68.
Ruben RJ. Redefining the survival of the fittest: communication disorders in the 21st century. Laryngoscope. 2000;110 2 Pt 1:241–245.
Shearer AE, Kolbe DL, Azaiez H, et al. Copy number variants are a common cause of nonsyndromic hearing loss. Genome Med. 2014;6:37.
Plevova P, Paprskarova M, Tvrda P, et al. STRC deletion is a frequent cause of slight to moderate congenital hearing impairment in the Czech Republic. Otol Neurotol. 2017;38:e393–e400.
Yokota Y, Moteki H, Nishio SY, et al. Frequency and clinical features of hearing loss caused by STRC deletions. Sci Rep. 2019;9:4408.
Kim NK, Kim AR, Park KT, et al. Whole-exome sequencing reveals diverse modes of inheritance in sporadic mild to moderate sensorineural hearing loss in a pediatric population. Genet Med. 2015;17:901–911.
Kim SY, Park G, Han KH, et al. Prevalence of p.V37I variant of GJB2 in mild or moderate hearing loss in a pediatric population and the interpretation of its pathogenicity. PLoS ONE. 2013;8:e61592.
Min BJ, Kim N, Chung T, et al. Whole-exome sequencing identifies mutations of KIF22 in spondyloepimetaphyseal dysplasia with joint laxity, leptodactylic type. Am J Hum Genet. 2011;89:760–766.
Mandelker D, Amr SS, Pugh T, et al. Comprehensive diagnostic testing for stereocilin: an approach for analyzing medically important genes with high homology. J Mol Diagn. 2014;16:639–647.
Kim BJ, Kim AR, Lee C, et al. Discovery of CDH23 as a significant contributor to progressive postlingual sensorineural hearing loss in Koreans. PLoS ONE. 2016;11:e0165680.
Kim BJ, Ueyama T, Miyoshi T, et al. Differential disruption of autoinhibition and defect in assembly of cytoskeleton during cell division decide the fate of human DIAPH1-related cytoskeletopathy. J Med Genet. 2019;56:818–827.
Lee SY, Joo K, Oh J, et al. Severe or profound sensorineural hearing loss caused by novel USH2A variants in Korea: potential genotype-phenotype correlation. Clin Exp Otorhinolaryngol. 2019. https://doi.org/10.21053/ceo.2019.00990 [Epub ahead of print].
Francey LJ, Conlin LK, Kadesch HE, et al. Genome-wide SNP genotyping identifies the Stereocilin (STRC) gene as a major contributor to pediatric bilateral sensorineural hearing impairment. Am J Med Genet A. 2012;158A:298–308.
Rozen S, Skaletsky H. Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol. 2000;132:365–386.
Vona B, Hofrichter MA, Neuner C, et al. DFNB16 is a frequent cause of congenital hearing impairment: implementation of STRC mutation analysis in routine diagnostics. Clin Genet. 2015;87:49–55.
Wesdorp M, Murillo-Cuesta S, Peters T, et al. MPZL2, encoding the epithelial junctional protein myelin protein zero-like 2, is essential for hearing in man and mouse. Am J Hum Genet. 2018;103:74–88.
Choi BY, Park G, Gim J, et al. Diagnostic application of targeted resequencing for familial nonsyndromic hearing loss. PLoS ONE. 2013;8:e68692.
Kim BJ, Kim AR, Park G, et al. Targeted exome sequencing of deafness genes after failure of auditory phenotype-driven candidate gene screening. Otol Neurotol. 2015;36:1096–1102.
Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424.
Oza AM, DiStefano MT, Hemphill SE, et al. Expert specification of the ACMG/AMP variant interpretation guidelines for genetic hearing loss. Hum Mutat. 2018;39:1593–1613.
Abou Tayoun AN, Pesaran T, DiStefano MT, et al. Recommendations for interpreting the loss of function PVS1 ACMG/AMP variant criterion. Hum Mutat. 2018;39:1517–1524.
Kim SY, Kim AR, Kim NK, et al. Strong founder effect of p.P240L in CDH23 in Koreans and its significant contribution to severe-to-profound nonsyndromic hearing loss in a Korean pediatric population. J Transl Med. 2015;13:263.
Delen D, Walker G, Kadam A. Predicting breast cancer survivability: a comparison of three data mining methods. Artif Intell Med. 2005;34:113–127.
Yildirim O, Plawiak P, Tan RS, et al. Arrhythmia detection using deep convolutional neural network with long duration ECG signals. Comput Biol Med. 2018;102:411–420.
Karim F, Majumdar S, Darabi H, et al. LSTM fully convolutional networks for time series classification. IEEE Access. 2018;6:1662–1669.
Zhang Y, Malekpour M, Al-Madani N, et al. Sensorineural deafness and male infertility: a contiguous gene deletion syndrome. J Med Genet. 2007;44:233–240.
Cooper TG, Noonan E, von Eckardstein S, et al. World Health Organization reference values for human semen characteristics. Hum Reprod Update. 2010;16:231–245.
Karger L, Khan WA, Calabio R, et al. Maternal uniparental disomy of chromosome 15 and concomitant STRC and CATSPER2 deletion-mediated deafness–infertility syndrome. Am J Med Genet A. 2017;173:1436–1439.
Kim BJ, Han JJ, Shin SH, et al. Characterization of detailed audiological features of cytomegalovirus infection: a composite cohort study from groups with distinct demographics. Biomed Res Int. 2018;2018:7087586.
Yu Y, Yang Y, Lu J, et al. Two compound heterozygous were identified in SLC26A4 gene in two Chinese families with enlarged vestibular aqueduct. Clin Exp Otorhinolaryngol. 2019;12:50–57.
Avidan N, Tamary H, Dgany O, et al. CATSPER2, a human autosomal nonsyndromic male infertility gene. Eur J Hum Genet. 2003;11:497–502.
Hoppman N, Aypar U, Brodersen P, et al. Genetic testing for hearing loss in the United States should include deletion/duplication analysis for the deafness/infertility locus at 15q15.3. Mol Cytogenet. 2013;6:19.
Sagong B, Baek JI, Bok J, et al. Identification of a nonsense mutation in the STRC gene in a Korean family with moderate hearing loss. Int J Pediatr Otorhinolaryngol. 2016;80:78–81.
Acknowledgements
This study was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (2018R1A2B2001054 to B.Y.C., 2018R1D1A1B07047966 to Seungmin Lee, 2017R1D1A1B03034401 to D.-Y.O., and 2018R1D1A1B07046159 to B.J.K.); a grant from the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (grant number HI17C0952 to B.Y.C.); and the research fund of Chungnam National University (B.J.K.). This document has been reviewed by professional editors who are native speakers of English (http://www.emendology.com/).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosure
The authors declare no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Kim, B.J., Oh, DY., Han, J.H. et al. Significant Mendelian genetic contribution to pediatric mild-to-moderate hearing loss and its comprehensive diagnostic approach. Genet Med 22, 1119–1128 (2020). https://doi.org/10.1038/s41436-020-0774-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-020-0774-9
Keywords
This article is cited by
-
MPZL2—a common autosomal recessive deafness gene related to moderate sensorineural hearing loss in the Chinese population
BMC Medical Genomics (2024)
-
The congenital hearing phenotype in GJB2 in Queensland, Australia: V37I and mild hearing loss predominates
European Journal of Human Genetics (2024)
-
Genetic screening of a Chinese cohort of children with hearing loss using a next-generation sequencing panel
Human Genomics (2023)
-
Frequency of the STRC-CATSPER2 deletion in STRC-associated hearing loss patients
Scientific Reports (2022)
-
Full etiologic spectrum of pediatric severe to profound hearing loss of consecutive 119 cases
Scientific Reports (2022)
