Kir6.2 as a Candidate Gene for Neonatal Diabetes
Studies in isolated cells and tissues, animal models, and human genetics have firmly established the central role of the KATP channel in insulin secretion and suggested that Kir6.2 is a good candidate gene for neonatal diabetes. First, overexpression in β-cells of a mutant Kir6.2 with a reduced ATP sensitivity caused mice to develop severe neonatal diabetes.[24] Second, the fact that loss-of-function mutations in Kir6.2 or SUR1 leads to CHI suggests that gain-of-function mutations in the same genes might cause diabetes. Gain- and loss-of-function mutations in the same ion channel gene have been shown to cause opposing phenotypes,[29] and hyperglycemia and hypoglycemia can result from inactivating and activating mutations, respectively, in glucokinase.[30] Third, as discussed more fully below, a common polymorphism in the Kir6.2 gene (E23K) is consistently associated with type 2 diabetes in both large-scale studies and meta-analyses,[31,32,33] so a severe mutation might cause monogenic diabetes.
Gloyn et al.[2] first reported that Kir6.2 mutations result in neonatal diabetes. They showed that heterozygous Kir6.2 mutations occurred in 10 of 29 patients diagnosed with diabetes before 6 months of age who required continuing insulin treatment after diagnosis. This study established that most Kir6.2 mutations were spontaneous, that some mutations were more prevalent, and that neurological features occurred in some patients. It also showed that the most common mutation, R201H, caused a reduced response of the KATP channel to ATP, consistent with a gain-of-function mutation, and that while patients with this mutation did not secrete insulin in response to glucose, they did respond to sulfonylureas. Subsequent studies have confirmed and extended these results.
Heterozygous mutations in Kir6.2 are the most common cause of neonatal diabetes in multiple populations and ethnic groups, accounting for 40-64% of cases in large series of permanent neonatal diabetes ( Table 1 ). To date, at least 63 patients with activating mutations in Kir6.2 have been described, comprising 21 different mutations in 49 families[2,34,35,36,37,38,39,40,41,42,43,44] (E.L. Edghill, S. Ellard, S. Flanagan, and A.T.H., personal communication). The distribution of these residues within the single exon KCNJ11 gene is shown in Fig. 1. Four mutations (V59M, R201H, R201C, and Y330C) have been described in more than one family, with the most common being R201H and V59M. Since most (>90%) mutations arose spontaneously, positions 59 and 201 represent recurrent mutations rather than founder mutations within populations. At position 201, there is a CpG dinucleotide, which represents a hot spot for mutations in eukaryote genes. In seven families, there was autosomal dominant inheritance from either the mother or father.[2,37,41,44] In a single family, there was a paternal germline mosaicism, resulting in the R201C mutation being detected in the leukocyte DNA of two half-siblings but not in that of the father.[35] The possibility of paternal germline mosaicism should therefore be considered when counseling parents of a child with an apparently de novo mutation of the risk of subsequent children being affected.
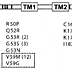
The location of mutations causing neonatal diabetes within the linear sequence of Kir6.2. The numbers in brackets give the number of patients carrying the indicated mutation (no number is given if this is 1). Residues that represent the two common hot spots for mutations are boxed (2,33-44 and E.L. Edghill, S. Ellard, S. Flanagan, and A.T.H., personal communication). N, NH2-terminal; C, COOH-terminal; TM1, first transmembrane domain; TM2, second transmembrane domain.
There is a spectrum of phenotypes associated with activating mutations in Kir6.2 ( Table 2 ). Almost all patients have neonatal diabetes, which may either be permanent or more rarely may remit (and hence be transient). Some patients have additional neurological features.
Neonatal Diabetes. The median age of diagnosis is 6 weeks, with all patients being diagnosed before 6 months of age, except for four cases diagnosed in childhood or early adult life ( Table 2 ). Most (79%) patients are diagnosed before 3 months, which has been suggested as a criterion for neonatal diabetes. A large Italian study of early-onset diabetes suggested that diagnosis by 6 months is the boundary between nonautoimmune diabetes and type 1 diabetes.[45] Consistent with this idea, Kir6.2 mutations were not detected in 101 patients with insulin-treated diabetes diagnosed between 6 and 24 months ( Table 1 ). This suggests that screening for Kir6.2 mutations is appropriate for all patients diagnosed before 6 months but not for patients diagnosed later than this.
The majority of patients have low birth weights (median 2.58 kg) with 71% below the 10th centile for gestational age in keeping with a lower fetal insulin production resulting in reduced insulin-mediated growth in utero. Patients show rapid catch-up growth after insulin treatment.
Most patients with Kir6.2 mutations present with symptomatic hyperglycemia with median blood glucose concentrations of 33.5 mmol/l, and in many cases there is ketoacidosis. However, the relatively late age of diagnosis in many cases suggests there is not absolute insulin deficiency from birth. Patients require insulin treatment from diagnosis; this is permanent in all but five cases where there is evidence of remission with insulin able to be completely discontinued at least for 1 year. These patients were defined clinically as having transient neonatal diabetes.[41,44]
The phenotype of diabetes due to Kir6.2 mutations differs from that found for other causes of neonatal diabetes. Permanent neonatal diabetes due to homozygous glucokinase mutations[46] or pancreatic aplasia due to homozygous IPF1 mutations[47,48] results in a more severe insulin deficiency as shown by a lower birth weight and a younger age at diagnosis. In keeping with pancreatic autoantibodies being rare in patients who are diagnosed with diabetes before 6 months,[45] no patients with Kir6.2 mutations have pancreatic autoantibodies, but these are found in patients with FOXP3 mutations leading to IPEX syndrome.[45] Transient neonatal diabetes resulting from the common abnormalities of the imprinted region on 6q24 differs from transient neonatal diabetes caused by Kir6.2 mutations. The former are born lighter (mean 2.1 vs. 2.6 kg) and diagnosed earlier (1.0 vs. 5.2 weeks), but remit earlier (16 vs. 76 weeks).[1,41]
In addition to neonatal diabetes, it is becoming clear that neurological features, particularly developmental delay, muscle weakness, and epilepsy, are associated with some Kir6.2 mutations.[2,34,35,37,39,42] The diabetes of these patients is similar to that found for patients without neurological features, but its management is more difficult due to marked communication problems and the risk that hypoglycemia can precipitate seizures in patients with epilepsy.
There has been some controversy about whether the neurological features associated with Kir6.2 mutations constitute a distinct syndrome or are a secondary consequence of diabetes or its treatment. There is now strong evidence in favor of the former. First, developmental delay is not a typical feature of neonatal diabetes from other causes.[1,46,49] Second, the pattern of neurological features is distinct from that resulting from cerebral edema, which may occur in the treatment of diabetic ketoacidosis, or as a consequence of severe hypoglycemia. Third, the presence of contractures at birth (arthrogryposis) in some patients is indicative of a neurological defect in utero.[2] Fourth, as discussed below, there is a strong genotype-phenotype relationship with the functional severity of the mutations in the heterozygous state correlating with differences in clinical phenotype. Finally, the neurological features are consistent with the tissue distribution of Kir6.2 in muscle, nerve, and brain.[3,9] Thus, we believe it appropriate to define a novel syndrome that is associated with some Kir6.2 mutations. We propose this be known as developmental delay, epilepsy, and neonatal diabetes (DEND) syndrome.[50]
Four patients (7%) have a very severe neurological phenotype that exhibits all the features of DEND syndrome.[2,42] They show marked developmental delay, motor weakness, and epilepsy in addition to diabetes. Two children died as a consequence of the neurological features within the first 15 months of life.[2,42] The other two are unable to talk, stand, or walk unaided at the ages of 5 and 18.[2] All four patients had generalized epilepsy before the age of 1. In all cases, electroencephalograms were abnormal with bilateral sharp waves, and two patients showed hypsarrhythmia. These patients also have mild dysmorphic features including a prominent metopic suture, bilateral ptosis, and a downturned mouth (the last two are both features of muscle weakness).
A less severe clinical picture, consisting of neonatal diabetes with developmental delay and/or muscle weakness but not epilepsy, is more common (14 of 51 patients)[2,34,35,37,39] ( Table 2 ). We refer to this as intermediate DEND syndrome. Motor milestones are delayed 1-2 years but less severely than in patients with epilepsy, and there is a variable degree of motor weakness. The most marked feature of social delay is the late development of speech, which may not occur until 5 years.
There is emerging evidence for a clear genotype-phenotype relationship for Kir6.2 mutations. Of the 24 patients with mutations at R201 described to date, all but 3 have nonremitting neonatal diabetes without neurological features ( Table 2 ). Conversely, 10 of 13 patients with the V59M mutation have developmental delay and features consistent with intermediate DEND syndrome ( Table 2 ). The mutations associated with full DEND syndrome are not found in less severely affected patients. A milder phenotype is associated with the C42R mutation; two of four patients did not develop diabetes until early adult life, one patient developed diabetes at age 3 years, and another exhibited transient neonatal diabetes.[44] However, although these data suggest a strong correlation between phenotype and genotype, the association is not absolute, as shown by differences in the degree to which β-cell function is impaired in patients with the R201H[2,38,43] and C42R[44] mutations and the absence of developmental delay in three patients with the V59M mutation. This implies that genetic background and environmental factors influence the clinical phenotype, as found for other monogenic subtypes of diabetes.[51,52]
The reason that some Kir6.2 mutations give rise to transient neonatal diabetes is unknown, but possible explanations include a reduced insulin requirement at the time of remission; compensation at the level of the β-cell, pancreas, or whole body that is able to overcome the effects of the channel defect; or changes in β-cell turnover due to the mutation in the gene encoding Kir6.2.
Figure 2 maps the mutations associated with neonatal diabetes onto a structural model of Kir6.2.[53] Residues associated with neonatal diabetes alone (transient neonatal diabetes or permanent neonatal diabetes) lie within the putative ATP-binding site (R50, I192, R201, and F333) or are located at the interfaces between Kir6.2 subunits (F35, F35, C42 and E332) or between Kir6.2 and SUR1 (G53). Mutations that cause additional neurological features occur at residues that lie at some distance from the ATP-binding site. Thus, Q52 sits within the cytosolic part of the NH2-terminal domain, which is thought to contribute to functional coupling of SUR1 to Kir6.2.[54,55] Residue V59 lies within the "slide helix," a domain implicated in the opening and closing of the pore.[53,56] C166 lies at the cytosolic end of the outer transmembrane helix close to the helix-bundle crossing suggested to form an inner gate to the channel,[57] and I296L lies within the permeation pathway at the mouth of the transmembrane pore in a region postulated to play a role in gating.[53,58]

A: Structural model of Kir6.2 (53) viewed from the side. For clarity, only two transmembrane domains and two separate cytosolic domains are shown. Residues mutated in permanent neonatal diabetes are shown in blue, in transient neonatal diabetes in green, and in DEND syndrome in red. ATP (purple) is docked into its binding site. B: Close-up of the putative ATP-binding site. Residues mutated in permanent neonatal diabetes are shown in blue, in transient neonatal diabetes in green, and ATP is shown in red.
All mutations studied to date produce a marked decrease in the ability of ATP to block the KATP channel when expressed in heterologous systems.[2,41,44,50,59,60] This reduction in ATP sensitivity means the channel will open more fully at physiologically relevant concentrations of ATP (1-5 mmol/l), leading to an increase in the KATP current. In pancreatic β-cells, an increase in KATP current will hyperpolarise the membrane, suppressing electrical activity, Ca2+ influx, and insulin secretion, and thereby causing diabetes.[48]
The molecular mechanism by which the ATP sensitivity of the Kir6.2 subunit is reduced varies between mutations. Studies to date suggest that most mutations associated with neonatal diabetes alone impair ATP-dependent channel inhibition without much change in the fraction of time the channel spends in the open state in the absence of ATP (the intrinsic open probability).[59,60] This is consistent with the fact that most of these mutations lie within the predicted ATP-binding site (Fig. 2). Mutations associated with neurological features, however, markedly bias the channel toward the open state, thus increasing the intrinsic open probability.[59,50,60] This indirectly reduces the ability of ATP to block the channel because ATP stabilizes the long-closed state of the channel, which is now less frequent.[61]
To date, most functional studies have been conducted in Mg2+-free solutions, as it is easier to study exactly how Kir6.2 mutations affect KATP channel ATP sensitivity in the absence of the stimulatory effects of Mg-nucleotides. However, it is also important to examine the effect of Kir6.2 mutations in the presence of Mg2+ because Mg2+ is always present in the cell and stimulation by Mg-nucleotides will shift the ATP sensitivity to higher ATP concentrations. Recent studies suggest that the reduction in KATP channel ATP sensitivity is greater in the presence of Mg2+ and that KATP currents recorded at physiological levels of MgATP (>1 mmol/l) are strikingly larger for DEND syndrome mutations than transient neonatal diabetes mutations, consistent with the severity of the clinical phenotype.[41,50,60] Thus, it appears that, in addition to reducing the inhibitory effect of ATP at Kir6.2, some mutations enhance MgATP stimulation mediated via SUR1,[50,60] perhaps by influencing interactions between Kir6.2 and SUR1. Finally, all experiments to date have been performed by expression in heterologous systems, such as Xenopus oocytes. Thus, studies using β-cells, neurons, and animal models are now needed.
The Importance of Heterozygosity. All neonatal diabetes-associated mutations are heterozygous, so both wild-type and mutant Kir6.2 are expressed in the same cell. Because Kir6.2 is a tetramer,[21] there will be a mixed population of channels, each containing between zero and four mutant subunits. The ATP sensitivity of any individual channel in this population will depend on the number of mutant subunits it contains and the extent to which each subunit contributes to the overall ATP sensitivity. This contribution may vary according to whether the mutation affects ATP binding or the intrinsic open probability. Binding of a single ATP molecule closes the KATP channel.[62] This means that if a mutation influences just ATP binding, only channels with four mutant subunits will have a markedly reduced ATP sensitivity. If wild-type and mutant Kir6.2 subunits distribute according to binomial theory, homomeric mutant channels will only account for one-sixteenth of channels in the heterozygous population; thus, the shift in ATP sensitivity will be small. Conversely, mutations that influence the intrinsic open probability will affect 15 of 16 channels (albeit to different extents), as each heteromeric channel will have at least one mutant subunit. Thus, the shift in ATP sensitivity will be larger. This may help explain why mutations that affect the intrinsic open probability produce a more severe clinical phenotype than those that affect the ATP-binding site. Studies are now required to determine whether this idea is correct and if wild-type and mutant subunits distribute as binomial theory predicts.
Diabetes. 2005;54(9):2503-2513. © 2005 American Diabetes Association, Inc.
Cite this: Activating Mutations in Kir6.2 and Neonatal Diabetes - Medscape - Sep 01, 2005.






Comments