
Abstract
Novel molecular mechanisms of the pathophysiology of heart failure (HF) are continuously being discovered, including epigenetic regulation. Among epigenetic marks, the role of DNA hypomethylation in shaping heart morphology and function in vivo and the pathogenesis of cardiomyopathy and/or HF, especially in adults, has not been clearly established. Here we show that the strong expression of DNA methyltransferase 1 (Dnmt1) is obviously downregulated in the WT adult rat heart with age. By contrast, the expression of Dnmt1 is upregulated suddenly in heart tissues from pressure overload-induced HF mice and adriamycin-induced cardiac injury and HF mice, consistent with the increased expression of Dnmt1 observed in familial hypertrophic cardiomyopathy (FHCM) patients. To further assess the role of Dnmt1, we generated myocardium-specific Dnmt1 knockout (Dnmt1 KO) rats using CRISPR-Cas9 technology. Echocardiographic and histopathological examinations demonstrated that Dnmt1 deficiency is associated with resistance to cardiac pathological changes and protection at the global and organization levels in response to pathological stress. Furthermore, Dnmt1 deficiency in the myocardium restricts the expressional reprogramming of genes and activates pathways involved in myocardial protection and anti-apoptosis in response to pathological stress. Transcriptome and genome-wide DNA methylation analyses revealed that these changes in regulation are linked to alterations in the methylation status of genes due to Dnmt1 knockout. The present study is the first to investigate in vivo the impact of genome-wide cardiac DNA methyltransferase deficiency on physiological development and the pathological processes of heart tissues in response to stress. The exploration of the role of epigenetics in the development, modification, and prevention of cardiomyopathy and HF is in a very preliminary stage but has an infinite future.
Similar content being viewed by others


Introduction
Heart failure (HF) is a complex pathophysiological syndrome that arises from a primary defect in the ability of the heart to take in and/or eject enough blood [1]. The clinical manifestations of HF are mainly the result of the primary myocardial disease, such as inherited dilated cardiomyopathy (DCM). Novel molecular mechanisms of HF pathophysiology include epigenetic regulation, such as by DNA methylation, the most common epigenetic modification in the mammalian genome [2,3,4].
In the process of DNA methylation, a methyl group is added to cytosine in the dinucleotide sequence CG [5]. The methylation of unmethylated CpG sites located in promoter regions leads to transcriptional inactivation of genes partly by suppressing transcription factor binding, and the methylation of intragenic regions also contributes to gene regulation and alternative splicing [5, 6].
Changes in DNA methylation have been shown to contribute to the pathogenesis of cardiovascular disease, including DCM and HF, by regulating gene expression [7,8,9]. A genome-wide study of DNA methylation in the hearts of end-stage cardiomyopathic patients showed that methylation is significantly reduced in the promoters of upregulated genes but unchanged in the promoters of downregulated genes [10, 11] The DNA methylation pattern in the myocardium is altered in DCM, and DNA methylation is responsible for the hypermutability of distinct cardiac genes [12,13,14]. Increased DNA methylation plays a causative role in programming heart hypertrophy and reducing cardiac contractility [9, 15]. These findings support possible role that DNA methylation in regulating the pathological process associate with HF.
The main enzymatic function of the maintenance DNA methyltransferase (DNMT) Dnmt1 is to faithfully restore methylation to newly formed daughter strands of DNA [16, 17]. Deletion of Dnmt1 is embryonically lethal, based on its essential role in the embryonic and primary development process of smooth muscle cells [18, 19], and consistent with this role, genomic DNA hypomethylation is observed in tissues affected by human gastrointestinal disease [20, 21]. In contrast to its family members Dnmt3a and Dnmt3b, Dnmt1 deficiency does not affect sarcomere assembly and cytotoxicity in embryonic cardiomyocytes [22, 23]. However, how DNA hypomethylation shapes heart morphology and function in vivo within the pathogenesis of cardiomyopathy and/or HF, especially in adults, remains unclear.
Therefore, to investigate the effect of Dnmt1 deficiency on heart development and the response to stress and its possible mechanisms, we generated a Dnmt1 conditional knockout (KO) rat and an α myosin heavy chain Cre (α-MHC-Cre) rat using CRISPR-Cas9 technology and selectively eliminated the methyltransferase Dnmt1 in the myocardium in α-MHC-Cre rats. Geometric and functional analyses of the heart revealed no differences between myocardium-specific Dnmt1 KO rats, referred to as Dnmt1-KO, and wild-type (WT) rats from birth to 12 months of age. However, rats with Dnmt1 deficiency exhibited resistance to cardiac pathological changes in response to pathological stress, with protection at the global, organizational and molecular levels. Furthermore, transcriptome and genome-wide DNA methylation analyses demonstrated that Dnmt1 deficiency in the myocardium inhibited the reprogramming of gene expression and activated pathways involved in myocardial protection and anti-apoptosis in response to pathological stress. These changes in the regulation of expression due to the Dnmt1 KO were related to the methylation status of genes.
These in vivo findings are the first to demonstrate the function and importance of Dnmt1 in the development of the heart and cardiac pathogenesis in DCM and HF.
Materials and methods
Animals
All rats we used in this paper were bred in standard cages in an Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC) accredited animal facility. All animal experiments were approved by Animal Care and Use Committees of the Institute of Laboratory Animal Science, Peking Union Medical College, China (ZLF18003).
Dnmt1 conditional knockout (Dnmt1 cKO) rats were established in our lab as previously reported [24]. In brief, a pair of synthetic oligonucleotides for sgRNA (sgRNA1: TAGGCGAGGGGCGGGACCGATG; sgRNA2: AAACCATCGGTCCCGCCCCTCG) were annealed and then cloned into the pUC57-sgRNA expression vector, and the floxed plasmid donor was cloned into the pGSI plasmid. Both the Cas9 and sgRNA expression plasmids were linearized and used as templates for in vitro transcription. Fertilized eggs from female SD rat donors were prepared as described previously [24], and a mixture of the donor vector (4 ng/μl), Cas9 mRNA (25 ng/μl), and sgRNAs (10 ng/μl each) was microinjected into both the cytoplasm and male pronucleus of the fertilized eggs. The injected zygotes were then transferred to pseudo pregnant SD rats, which then carried to parturition.
α-MHC-Cre transgenic rats were established in our laboratory as previously reported [25]. In brief, the gene encoding Cre recombinase was inserted downstream of the α-MHC promoter, and the resulting plasmid was named pα-MHC-Cre. The plasmid was then linearized with NotI restriction endonuclease and purified for microinjection. Animals carrying the α-MHC-Cre transgene were identified using PCR analysis of tail genomic DNA using Cre-specific primers as previously reported [25]. The positive transgenic rats would be then maintained through mating with SD rats.
To specifically knockout Dnmt1 in the myocardium, we used the Cre-loxP system by crossing α-MHC-Cre rats with Dnmt1 cKO rats. Dnmt1+/− offspring positive for the α-MHC-Cre transgene (double-positive) were selected. In the second round of crossbreeding, the double-positive rats were crossed with Dnmt1 cKO rats, and Dnmt1−/− offspring positive for the α-MHC-Cre transgene were obtained as myocardium-specific Dnmt1-KO rats, referred to as Dnmt1-KO rats in this study.
Adriamycin treatment
Two-month-old KO rats and their WT littermates were used for Adriamycin (ADR) treatment. In the KO-ADR and WT-ADR groups, ADR was injected intraperitoneally in a constant volume of saline at 2.5 mg/kg every other day for a total of 2 weeks as previously reported [26, 27]. The groups designated as KO-saline and WT-saline received the same volume of saline. Echocardiography was performed on all rats on day 0 (the day before ADR treatment). All surviving rats were subjected to follow-up echocardiography at 3 months of age (2 weeks after the cessation of ADR treatment) and used for subsequent analysis.
Thoracic aorta constriction treatment
Thoracic aorta constriction (TAC) was carried out in 2-month-old C57BL6 mice as previously described [26, 28]. Wild-type (WT) mice were anesthetized with tribromoethanol at a concentration of 216 mg/kg body weight. The operation was carried out using a ventilator to ensure passive respiration. The aorta was constricted with a calibrator by employing a 27-gauge blunted needle. Sham surgery was performed identically without constricting the aorta. Expression analysis will be performed two months after treatment.
Human cardiac samples
Cardiac samples of left ventricular (LV) interventricular septa from six patients with hypertrophic cardiomyopathy who underwent morrow septal myectomy were used for expression analysis. To ensure maximum control of potential confounding effects, we chose patients without congenital heart disease, congestive HF, myocardial infarction or valvular heart diseases. Normal control samples were obtained from the same cardiac region in three healthy donors who died in accidents. The cardiac samples were always stored in liquid nitrogen until use. Written informed consents have been obtained either from participants or their relatives. Study protocol has been approved by Ethics Committee of Fuwai Hospital.
Genomic DNA preparation and genotyping
Genomic DNA was extracted by phenol/chloroform and recovered by alcohol precipitation from the tail tissue of 7-day-old rats. Each rat tail tissue sample was dissolved in SNET buffer (20 mM Tris-Cl, 400 mM NaCl, 5 mM EDTA, 1% SDS, 0.4 mg/mL proteinase K) for more than 4 h (55 °C). The lysate was then extracted, using equal volume of phenol/chloroform, which followed by equal volume chloroform. Aaqueous phase will be recovered. 2.5 volumes of alcohol were added to precipitate the genomic DNA, which was then dissolved in TE buffer (10 mM Tris/HCl, pH 8, 0.1 mM EDTA). For genotyping of genetically modified rats, then PCR primers were designed as previously reported [24, 25].
Survival analysis
The cumulative percentage mortality for all rats was counted every week from 0 day to the end of observation (2 weeks after the cessation of ADR treatment) as previously described [26]. Upon the death of each rat, postmortem analysis was conducted by a pathologist, and any changes in heart morphology and pathology were recorded. We then constructed Kaplan–Meier curves, with the log-rank test for survival analysis (SPSS 16.0 software).
Echocardiography
Echocardiographic inspection was carried out with an echocardiography system with a 16 MHz transducer (Vevo770, Canada). Rats were first anesthetized by isoflurane (1.5–2.5%). Normal body temperature of the rats was always maintained. Measurements were recorded as the mean value of at least three continuous cardiac cycles. Left ventricular (LV) internal diameter at end systole (LVEDS), LV internal diameter at end diastole (LVEDD) and LV anterior wall thickness at end systole (LVAWS) were obtained.
Histological analysis
For light microscopy, heart tissues were first fixed in formaldehyde (4%) and then mounted in paraffin blocks. Hematoxylin and eosin (H&E) or Masson’s trichrome staining was performed as previously described, and stained sections were examined using Aperio ImageScope v8.2.5 software [26, 27]. For transmission electron microscopy (TEM) analysis, heart tissues were routinely fixed in glutaraldehyde (2.5%) in phosphate buffer (0.1 M), then post fixed in osmium tetroxide buffer (1%, for 1 h). After staining with uranyl acetate and lead citrate, the sections were finally analyzed under a JEM-1230 transmission electron microscope as previously described [27].
Protein extraction and immunoblotting
Nuclear protein lysates of heart tissues were prepared following the manufacturer’s instructions (Thermo Fisher, 78833). The proteins were separated by SDS-PAGE. Then will be transferred to nitrocellulose membranes. The membranes were incubated with monoclonal antibodies against Dnmt1 (Thermo Fisher, MA5-16169, 1:1000). Antibody binding was detected with HRP-conjugated immunoglobulin G and a chemiluminescence system. Nucleolin was used for normalization (Abcam, ab22758, 1:1000). We used Image J software for Quantitative analysis of bands.
Immunofluorescence
Sections of heart tissues were prepared following standard pathological procedures as previously described [29]. The sections were dewaxed, then rehydrated, last epitopes were unmasked. The sections were then blocked and incubated with anti-Dnmt1 antibody (Thermo Fisher, MA5-16169, 1:50) and cTnT antibody (Abcam, ab45932, 1:200) overnight at 4 °C. Following washing with PBS, the sections were incubated with Alexa 555-conjugated goat anti-mouse IgG (Thermo Fisher, A-21424, 1:100) and Alexa 488-conjugated goat anti-rabbit IgG (Thermo Fisher, A-11008, 1:100) secondary antibodies for 1 h at room temperature. After washing with PBS and mounting in ProLong Gold anti-fade reagent with DAPI (Thermo Fisher, P36966), images of the sections were acquired by confocal laser scanning microscopy (Leica TCS SP2, Germany) and analyzed.
RNA-seq data analysis
Total RNA was isolated from heart tissues using TRIzol Reagent following the standard procedure. Library preparation, Illumina RNA sequencing, and data processing were performed as described previously [29, 30]. We use edgeR R package (3.12.1) for differential expression analysis. We first filtered out genes with RPKM < 1 in comparison. Multiple testing errors were corrected by the false discovery rate. Differentially expressed genes (DEGs) will be indicated by differences >2-fold. P values were then adjusted using the Benjamini–Hochberg method.
Genome-wide DNA methylation analysis
DNA methylation was analyzed by sodium bisulfite DNA sequencing as previously described [31, 32]. Genomic DNA was isolated with TIANamp Genomic DNA kit (Tiangen Biotech, Beijing, China). Then converted with EZ DNA Methylation Gold kit (Zymo Research). The primers for sodium bisulfite DNA sequencing were designed using online tools from MethPrimer, and the PCR products were cloned into the pEASY-T1 cloning vector (TransGen Biotech).
Statistical analysis
Data were analyzed with unpaired two-tailed Student’s t tests for two groups. One-way analysis of variance (ANOVA, with Tukey correction) was performed for multiple groups. The data are expressed as means ± SD. Differences were considered significant at P < 0.05.
Results
Dnmt1 is expressed in the myocardial nucleus and is induced under cardiomyopathy and HF
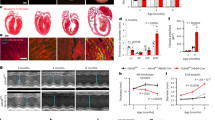
First, we tested the expression characteristics of Dnmt1 in heart tissues from WT rats. Immunofluorescence analysis revealed that Dnmt1 was localized primarily in the nucleolus, including cardiomyocytes and non-cardiomyocytes, with only a fraction in the cytoplasm (Fig. 1a). We then tested the expression features of Dnmt1 over time and found that Dnmt1 expression was strongest in the hearts of WT rats at 2 weeks of age. Interestingly, the expression was decreased thereafter with age (Fig. 1b, c).

a Immunofluorescence detection of Dnmt1 in the heart tissues of WT rats (scale bar = 100 μm). b Heart sampled from rat at 2 weeks, 1 month, 3 months, 5 months, 7 months and 10 months of age, and Dnmt1 expression was detected using western blot. d Dnmt1 expression in heart tissues of ADR treated and TAC mice models. f Dnmt1 expression in heart tissues from HCM patients and normal controls. c, e, g Quantitative analysis of Dnmt1 expression using Nucleoli for normalization.
In contrast to the decrease in Dnmt1 expression in normal adult hearts, Dnmt1 expression was increased obviously in the Adriamycin (ADR)-induced DCM/HF mouse model and pressure overload-induced (TAC treatment) hypertrophic cardiomyopathy (HCM)/HF mouse model (Fig. 1d, e, n = 3 mice per group, P < 0.01 vs. the saline or sham group). An analysis of the expression characteristics of Dnmt1 in HCM patients and healthy heart donors revealed that Dnmt1 expression was also increased significantly under cardiomyopathy in human heart tissues (Fig. 1f, g, n = 3 normal and n = 6 HCM patient samples, P < 0.001 vs. normal).
The abnormally increased expression of Dnmt1 under pathological states prompted us to further investigate the function of Dnmt1 in the myocardium, especially in response to stress.
The establishment of the cardiac-specific Dnmt1 knockout rat and survival rate observation in response to ADR-induced pathological stress
To explore how Dnmt1 shapes heart morphology and function in vivo within the pathogenesis of cardiomyopathy and/or HF, we selectively eliminated Dnmt1 from the α-MHC rat genome by crossing Dnmt1 cKO rats with α-MHC-Cre rats (Fig. 2a). Genotyping was carried out by PCR (Fig. 2b), and protein expression levels were further confirmed by western blot (Fig. 2c, d, n = 3 rats per group). In both pups and adults, there were no differences in appearance or status between WT rats and the cardiac-specific Dnmt1 KO (referred to as KO) rats.

a Breeding diagram of the genetically modified rats. b Gel electrophoresis of the genotyping. c Dnmt1 expression in heart tissues of Dnmt1 KO rats. d Quantitative analysis of Dnmt1 expression using Nucleoli for normalization. e Cumulative percentage mortality for WT-saline, WT-ADR, KO-saline and KO-ADR rats was calculated weekly from 0 to 4 weeks from the treatment of ADR.
Subsequently, we treated the KO rats with ADR to assess their response to stress. Cumulative rat mortality data of the KO and WT groups treated with ADR or saline (referred to as KO-ADR, WT-ADR, KO-saline and WT-saline) were recorded from day 0 of ADR treatment to the end of observation (2 weeks after the cessation of ADR treatment). Dilation and mural thrombi were observed in the hearts of rats during the postmortem examinations. The survival rate until the end of observation was 100% in the WT-saline (n = 8) and KO-saline (n = 6) group but only 75% in the WT-ADR group (Fig. 2e, P = 0.143, WT-ADR (n = 8) vs. WT-saline (n = 8) group). However, the survival rate in the KO-ADR group increased by 25% compared with the WT-ADR group (Fig. 2e, P = 0.205, KO-ADR (n = 6) vs. WT-ADR (n = 8) group). Thus, Dnmt1 KO to some extent increased the survival situation although not significantly by statistical analysis.
Dnmt1 knockout in the myocardium improves cardiac geometry and dysfunction in response to ADR-induced pathological stress
After the observation of survival analysis with the Dnmt1 deficiency when facing ADR. We next examined rat cardiac geometry and function in the WT-saline, KO-saline, WT-ADR and KO-ADR groups by echocardiography (Fig. 3a–e).

a The M-mode echocardiomyopathy screenshot of WT-saline, WT-ADR, KO-saline and KO-ADR rats at 2 weeks after cessation of ADR treatment. b Ratio of heart weight/body weight for four groups. c–e Echocardiographic parameters of Left ventricle (LV) diameter at end systole (LVEDS), LV anterior wall thickness at end systole (LVAWS) and LV fractional shortening (LVFS) for four groups at 2 weeks after cessation of ADR treatment.
We found that Dnmt1 KO reduced cardiac geometry disruption and dysfunction in the ADR-induced DCM/HF model. The DCM/HF phenotype was obviously induced by ADR treatment in the WT-ADR group, as demonstrated by the increased heart weight to body weight (HW/BW) ratio (Fig. 3b, n = 6, P < 0.05 vs. WT-saline), increased Left ventricle (LV) diameter at end systole (LVEDS, Fig. 3c, n = 6, P < 0.05 vs. WT-saline) and decreased LV anterior wall thickness at end systole (LVAWS, Fig. 3d, n = 6, P < 0.05 vs. WT-saline) at the end of observation (2 weeks after cessation of ADR treatment). Cardiac function was also impaired in the WT-ADR group, as demonstrated by the decreased LV fractional shortening (LVFS, Fig. 3e, n = 6, P < 0.05 vs. WT-saline) at the end of observation.
The changes in function and geometry induced by ADR were obviously ameliorated in the KO-ADR group compared with the WT-ADR group. HW/BW (Fig. 3b, n = 6, P = 0.384 vs. KO-saline), LVEDS (Fig. 3c, n = 6, P = 0.224 vs. KO-saline), LVAWS (Fig. 3d, n = 6, P = 0.053 vs. KO-saline) and LVFS (Fig. 3e, n = 6, P = 0.223 vs. KO-saline) did not significantly differ between the KO-ADR and KO-saline groups. In addition, we found that the ventricular cavities of KO-saline rats were slightly larger and ventricular wall thickness were slightly thinner, but there were no differences in echocardiographic parameters between the WT-saline and KO-saline groups.
The LVEDS, LVAWS and LVFS values of the WT-saline, KO-saline, WT-ADR and KO-ADR groups at the end of observation are shown in supporting data Table S1.
Dnmt1 knockout in the myocardium improves cardiac pathological changes in response to ADR-induced pathological stress
After assessing changes in cardiac geometry and function, pathological changes in response to ADR-induced stress were further detected in the WT-saline, KO-saline, WT-ADR and KO-ADR groups of rats.
Histopathological changes in gross morphology and myocardial microstructure were assessed by hematoxylin and eosin (H&E) staining. The thin wall, dilated chamber and malalignment in WT-ADR rats were obviously improved by Dnmt1 KO in the KO-ADR group (Fig. 4a, b), consistent with the gross morphology analysis by echocardiography. Moreover, collagen accumulation in the interstitial space, which was detected by Masson’s trichrome staining, increased obviously in the heart tissues of WT-ADR rats, and this increase was improved significantly in the heart tissues of KO-ADR rats (Fig. 4c). There were no obviously changes in KO-saline rats compared with that of WT-saline rats from H&E and Masson’s trichrome staining.

a Representative photographs of the whole-heart longitudinal sections with H&E staining from four groups at 2 weeks after cessation of ADR treatment. b Magnification of the left ventricle sections with H&E-stain (magnification ×400). c Magnification of Masson trichrome-stained left ventricle section (magnification ×400). d TEM analysis of sarcomeres and mitochondria of left ventricular free walls from four groups at 2 weeks after cessation of ADR treatment.
We subsequently detected morphological changes in the ultrastructure of myocytes in ventricles by electron microscopy. Swollen mitochondria, poorly organized myocardium and myocardiolysis were all obviously lessened by Dnmt1 KO in the KO-ADR group compared with the WT-ADR group (Fig. 4d). We found that a slight misalignment of the myocardial fibers in KO-saline rat group, but there was no significant difference between the KO-saline and WT-saline groups.
Dnmt1 knockout in the myocardium inhibits the reprogramming of gene expression in response to ADR-induced pathological stress
To obtain a complete understanding of the changes in gene expression in Dnmt1-KO rats in response to ADR-induced pathological stress, we performed transcriptomic analysis of the heart tissues of Dnmt1-KO rats as well as their WT littermates with and without ADR treatment. The mapping efficiency of the sequencing reads was >80%, and the samples clearly clustered by treatment (data not shown).
Differential expression analysis by EdgeR software revealed that compared to WT-saline rats, 1147 genes exhibited an increase in expression of at least twofold in WT-ADR rats, and 1193 genes exhibited reductions of expression of at least half. By contrast, in the KO-ADR group, 385 genes were upregulated and 415 genes were downregulated compared with the KO-saline group (Fig. 5a). These results indicate that Dnmt1 KO increases the resistance of rats to remodeling of gene expression in response to stimuli.

a Expression of genes changed at least twofold in both WT-ADR vs. WT-Saline and KO-ADR vs. KO-saline. b Five signaling pathways from KEGG pathway enrichment analysis in both WT-ADR vs. WT-Saline and KO-ADR vs. KO-saline. c mRNA levels of key molecules associate with myocardium structure and functions. d Heat map for mRNA levels of the key molecules from four groups at 2 weeks after cessation of ADR treatment.
Dnmt1 knockout in the myocardium activates pathways involved in myocardial protection and anti-apoptosis in response to ADR-induced pathological stress
Gene ontology (GO) enrichment analysis of the DEGs was carried out using Goseq. Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis was performed using KOBAS. The GO and KEGG analyses revealed that many pathways related to cell growth and development, cell morphology, cell organization and assembly, and cell signaling were altered in the KO-saline group compared with the WT-saline group (Table 1). In particular, many pathways involved in cardiac morphology and function and cardiac protection were significantly enriched (Table 1). The enrichment of these pathways suggests that the Dnmt1 KO altered gene expression.
ADR can induce myocardial damage and death, and the main mechanism is thought to be ROS. Activation of the MAPK and PI3K-Akt signaling pathways appears to be advantageous for damaged or failing myocardium. KEGG pathway enrichment analysis revealed that the changes in these two pathways in response to ADR treatment were more significant for KO rats than for WT rats (Fig. 5b, P = 0.0835 for KO-ADR vs. KO-saline, P = 0.1541 for WT-ADR vs. WT-saline for the MAPK signaling pathway; P = 0.0001 for KO-ADR vs. KO-saline, P = 0.0207 for WT-ADR vs. WT-saline for the PI3K-Akt signaling pathway). AMPK is supposed to play an important role in cellular energy homeostasis and is usually activated in energy deficiency. This pathway changed significantly in WT but not KO rats treated with ADR (Fig. 5b, P = 0.0099 for WT-ADR vs. WT-saline; P = 0.0284 for KO-ADR vs. KO-saline). Adrenergic signaling, which can reflect the status of stress in the myocardium, including ischemia and injury, was also altered significantly in WT but not KO rats in response to the ADR stimulus (Fig. 5b, P = 0.0025 for WT-ADR vs. WT-saline; P = 0.4132 for KO-ADR vs. KO-saline). In addition to these functional pathways, pathways involved in structure exhibited differences in regulation between KO and WT rats treated with ADR. For example, the enrichment of tight junctions was more significant in KO than in WT rats in response to the stimulus (Fig. 5b, P = 0.00685 for WT-ADR vs. WT-saline; P = 0.4955 for KO-ADR vs. KO-saline).
We then determined the mRNA levels of the key molecules in the crucial pathways discussed above in WT and KO rats facing stress. Compared with the WT-saline group, the WT-ADR group exhibited remarkable changes in key molecules involved in the cellular response to environmental stresses and the control of a variety of cell functions, including apoptosis and proliferation, such as Mapk10, Map3k7, Akt2, Bcl2, Pdk2, and Casp4. In addition, obvious changes in key molecules involved in myocardium structure and functions such as cytoskeleton regulation and contraction were observed, including Nppa, Tnni1, Myl7, Myh6, Myh10, Tnnt3, and Col1α1. By contrast, the changes in the mRNA levels of these key molecules in response to stimuli were significantly reduced in KO rats (Fig. 5c, d). The FRKM (Fragments per kilobase of transcript sequence per millions base pairs sequenced) values of all mRNAs in WT and KO rats after ADR treatment are presented in supporting data Tables S2 and S3.
In summary, the biofunction pathways affected in rats facing stress were associated with changes in survival phenotypes, cardiac geometry and function, and pathology.
Changes in the regulation of gene expression due to Dnmt1 knockout are related to methylation status
We identified 3626 genes with differentially methylated regions (DMRs) between KO and WT rats with at least one CpG site with significant demethylation. In rats treated with ADR, this number increased to 17,800. Dnmt1 loss induced global genomic CpG hypomethylation. We analyzed the distribution characteristics of the DMRs and observed that some CpG sites were strongly hypermethylated in KO rats, but these sites were considerably outnumbered by the number of strongly hypomethylated CpG sites (Fig. 6a). Similar patterns were not observed for genomic CHG or CHH (H can be A, C, or T) sites (Supplementary Fig. S1).

a Differentially methylated regions (DMRs) for CG DMR gene region distribution in both KO-saline vs. WT-saline and KO-ADR vs. WT-ADR. b Average of CpG methylation changes between KO-saline and WT-saline groups. c Average of CpG methylation changes between KO-ADR and WT-ADR groups. d Correlation analysis between transcriptional level and methylation state for gene with significant changes in transcriptome analysis. e Six signaling pathways from KEGG pathway enrichment analysis in both WT-ADR vs. WT-Saline and KO-ADR vs. KO-saline.
Across the KO rat genome, there was 20–30% loss of CpG methylation, with reductions of the average CpG methylation levels of a majority of genes from 0.60–0.65 to 0.4–0.35 compared with WT rats (Fig. 6b). However, this average decreased from 0.85–0.90 to 0.55–0.60 in KO rats compared with WT rats after ADR treatment (Fig. 6c).
We found that the remodeling of gene expression at the transcriptional level was correlated with methylation state. Among the 2340 genes whose expression was altered by at least twofold in WT-ADR rats compared with WT-saline rats (1147 genes with increased expression and 1193 with decreased expression), 1034 exhibited significant differences in methylation at one or more sites, including introns, exons, and promoters (Fig. 6d). These results indicate that the reduced remodeling of gene expression in KO rats in response to ADR-induced pathological stress was due to Dnmt1 deficiency.
KEGG pathway enrichment analysis revealed that the most overrepresented biological process categories in the groups treated with ADR were those associated with the ribosome, glycolysis, the HIF-1 signaling pathway and metabolic pathways. Specifically, the adrenergic signaling (Fig. 6e, P = 0.4173 for WT-ADR vs. WT-saline; P = 0.5510 for KO-ADR vs. KO-saline), PI3K-Akt (Fig. 6e, P = 0.2697 for WT-ADR vs. WT-saline; P = 0.4642 for KO-ADR vs. KO-saline), MAPK (Fig. 6e, P = 0.2471 for WT-ADR vs. WT-saline; P = 0.7111 for KO-ADR vs. KO-saline), apoptosis (Fig. 6e, P = 0.2291 for WT-ADR vs. WT-saline; P = 0.5722 for KO-ADR vs. KO-saline), citrate cycle (Fig. 6e, P = 0.0693 for WT-ADR vs. WT-saline; P = 0.3722 for KO-ADR vs. KO-saline) and AMPK (Fig. 6e, P = 0.0084 for WT-ADR vs. WT-saline; P = 0.3900 for KO-ADR vs. KO-saline) networks were predicted to be inhibited based on the gene expression patterns, indicating that KO rats may be less responsive to external stimuli than WT rats. The results were consistent with what we found in the transcriptome analysis. In particular, pathways regulating metabolism were enriched (Fig. 6e, P = 0.0984 for WT-ADR vs. WT-saline; P = 0.0060 for KO-ADR vs. KO-saline). These results suggest that the methyltransferase Dnmt1 may play an important role in the regulation of myocardial metabolism.
Discussion
The roles of genetics and epigenetics in the disease phenotype of HF in human patients are an exciting new frontier in cardiovascular medicine. In particular, knowledge of the impact of DNMTs, especially Dnmt1, on physiological and pathogenic cardiovascular processes remains very limited.
In this paper, we first generated a myocardium-specific Dnmt1 knockout rat using CRISPR-Cas9 technology. This knockout allowed us to specifically study the effects of Dnmt1 deficiency on heart tissues and exclude interference caused by other tissues. Geometric and functional observations of the heart based on echocardiographic and histopathological analysis revealed no differences between Dnmt1-KO and WT rats at birth up through 12 months of age. We subsequently investigated the response of Dnmt1-KO rats to pathological stress induced by ADR, a drug that exhibits dose-dependent toxicity and consequently induces cardiotoxicity, cardiomyopathy and even HF. Interestingly, rats with Dnmt1 deficiency exhibited reduced cardiac pathological changes and protection at the global, organizational and molecular levels in response to pathological stress. Furthermore, Dnmt1 deficiency in the myocardium resulted in reduced reprogramming of gene expression and the activation of pathways involved in myocardial protection and anti-apoptosis in response to pathological stress. Finally, transcriptome and genome-wide DNA methylation analyses showed that the changes in the regulation of gene expression were related to differences in the methylation status of genes due to the Dnmt1 knockout.
DNA methylation patterns are maintained by DNMTs, and our understanding of the roles of DNMTs in cardiac development and disease is in its early stages. Dnmt1 primarily methylates hemimethylated DNA, whereas Dnmt3a and -3b are de novo DNMTs that primarily methylate unmethylated DNA [5, 33]. Deletion of Dnmt1 or 3b leads to embryonic lethality [19, 33]. Dnmt3a KO mice appear normal at birth but die of unknown causes at 4 weeks of age, whereas Dnmt3b KO mice die between Embryo (E)13.5 and E16.5 due to abnormal heart structure [19, 34]. Cardiomyocyte-specific loss of Dnmt3b in adult mice induced by tamoxifen for 3–4 weeks, results in compromised systolic function, increased interstitial fibrosis, and myosarcoma disarray [35]. Phenotype analyses have revealed that Dnmt3a heterozygous KO mice have reduced heart rate variability, which is a predictor of adult HF [36, 37]. Dnmt3a is downregulated in the heart in patients with Tetralogy of Fallot, a condition associated with DNA hypomethylation [38, 39]. Dnmt3a is overexpressed in activated cardiac fibroblasts, leading to cardiac fibrosis [40, 41]. Dnmt3a knockdown in embryonic mouse cardiomyocytes plays an important role in regulating embryonic cardiomyocyte gene expression, morphology and function [22, 42].
Previous studies in mice have demonstrated that Dnmt1 is essential for embryo development and that Dnmt1 deficiency is lethal. However, cells treated with Dnmt1 siRNA do not exhibit abnormal morphology [22, 43]. Therefore, we speculated that the effects of Dnmt1 on the cardiovascular system are not evident in the embryonic stage. To observe the effects of Dnmt1 on myocardium development, studies of adult animals under both physiological and pathological conditions are needed. Our results showed that Dnmt1 deficiency in the myocardium has no obvious effects on the heart under physiological conditions and instead increases injury resistance and protection in the myocardium [22, 44, 45] under pathological conditions.
In addition, we performed the sequencing analysis on the myocardial tissue mixture, and we did not separate the cardiomyocytes and non-cardiomyocytes. Therefore, the obtained results showed that the significance of changes in myocardial protection and anti-apoptosis-related signaling pathways was much lower than what we predicted.
Based on our findings and those of others, inhibition of DNA methylation is a potential therapy for preventing cardiac injury and even treating HF [15, 46, 47]. Studies of the dynamics of epigenetic modifications will provide more accurate evidence of the roles of changing environmental factors in drug responses, thereby linking the environment with the genome and revealing potential strategies for reactivating silenced genes. Targeting methyltransferases may offer a possible therapeutic strategy for HF, detailed and in-depth studies must be carried out in subsequent projects. Additional research is obviously required to clarify how epigenetic mechanisms affect the onset and development of heart disease and heart regeneration to identify new drug targets for HF and to allow disease classification and risk stratification.
The present study is the first to investigate in vivo the impact of genome-wide cardiac DNMT deficiency on physiological development and the pathological process of heart tissues in response to stress. Knowledge of epigenetics and its relevance to the development, modification, and prevention of cardiomyopathy and HF disease is in a very preliminary stage but shows great potential.
References
Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Blaha MJ, et al. Executive summary: heart disease and stroke statistics—2014 update: a report from the American Heart Association. Circulation. 2014;129:399–410.
Saxonov S, Berg P, Brutlag DL. A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters. Proc Natl Acad Sci USA. 2006;103:1412–7.
Deaton AM, Adrian B. CpG islands and the regulation of transcription. Gene Dev. 2011;25:1010–22.
Gasiūnienė M, Zentelytė A, Wojtas B, Baronaitė S, Krasovskaja N, Savickienė J, et al. DNA methyltransferases inhibitors effectively induce gene expression changes suggestive of cardiomyogenic differentiation of human amniotic fluid‐derived mesenchymal stem cells via chromatin remodeling. J Tissue Eng Regen Med. 2019;13:469–81.
Kass SU, Pruss D, Wolffe AP. How does DNA methylation repress transcription? Trends Genet. 1997;13:444–9.
Flores K. Genome-wide association between DNA methylation and alternative splicing in an invertebrate. Bmc Genomics. 2012;13:480.
Gilsbach R, Preissl S, Grüning BrA, Schnick T, Burger L, Benes V, et al. Dynamic DNA methylation orchestrates cardiomyocyte development, maturation and disease. Nat Commun. 2014;5:5288.
Chamberlain AA, Mingyan L, Lister RL, Maslov AA, Yidong W, Masako S, et al. DNA methylation is developmentally regulated for genes essential for cardiogenesis. J Am Heart Assoc. 2014;3:e000976.
Stenzig J, Schneeberger Y, Löser A, Peters BS, Schaefer A, Zhao RR, et al. Pharmacological inhibition of DNA methylation attenuates pressure overload-induced cardiac hypertrophy in rats. J Mol Cell Cardiol. 2018;120:53–63.
Mehregan M, Mun-Kit C, Knowles DA, Lina C, Syed H, Thomas D, et al. Distinct epigenomic features in end-stage failing human hearts. Circulation. 2011;124:2411.
Mathiyalagan P, Keating ST, Du XJ, El-Osta A, et al. Interplay of chromatin modifications and non-coding RNAs in the heart. Epigenetics. 2014;9:101–12.
Whayne TF. Epigenetics in the development, modification, and prevention of cardiovascular disease. Mol Biol Rep. 2015;42:765–76.
Meurs KM, Kuan M. Differential methylation of CpG sites in two isoforms of myosin binding protein C, an important hypertrophic cardiomyopathy gene. Environ Mol Mutagen. 2011;52:161–4.
Yang Q, Sanbe A, Osinska H, Hewett TE, Klevitsky R, Robbins J. In vivo modeling of myosin binding protein C familial hypertrophic cardiomyopathy. Circ Res. 1999;85:841–7.
Daliao X, Chiranjib D, Man C, Kangling Z, John B, Zhice X, et al. Inhibition of DNA methylation reverses norepinephrine-induced cardiac hypertrophy in rats. Cardiovasc Res. 2014;101:373–82.
Marie-France R, Steves M, Normand B, France G, Chute IC, Annie B, et al. DNMT1 is required to maintain CpG methylation and aberrant gene silencing in human cancer cells. Nat Genet. 2003;33:61–65.
Omura N, Goggins M. Epigenetics and epigenetic alterations in pancreatic cancer. Int J Clin Exp Pathol. 2009;2:310–26.
Lei H, Oh SP, Okano M, Jüttermann R, Goss KA, Jaenisch R, et al. De novo DNA cytosine methyltransferase activities in mouse embryonic stem cells. Development. 1996;122:3195–205.
Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99:247–57.
Jorgensen BG, Berent RM, Ha SE, Horiguchi K, Sasse KC, Becker LS, et al. DNA methylation, through DNMT1, has an essential role in the development of gastrointestinal smooth muscle cells and disease. Cell Death Dis. 2018;9:474.
Kim GH, Ryan JJ, Archer SL. The role of redox signaling in epigenetics and cardiovascular disease. Antioxid Redox Sign. 2013;18:1920–36.
Fang X, Poulsen RR, Wang-Hu J, Shi O, Calvo NS, Simmons CS, et al. Knockdown of DNA methyltransferase 3a alters gene expression and inhibits function of embryonic cardiomyocytes. Faseb J. 2016;30:3238–55.
Tsai YT, Chang CM, Wang JY, Hou MF, Wang JM, Shiurba R, et al. Function of DNA methyltransferase 3a in lead (Pb(2+))-Induced Cyclooxygenase-2 gene. Environ Toxicol. 2015;30:1024–32.
Yuanwu M, Xu Z, Bin S, Yingdong L, Wei C, Jing M, et al. Generating rats with conditional alleles using CRISPR/Cas9. Cell Res. 2014;24:122–5.
Ma Y, Yu L, Pan S, Gao S, Chen W, Zhang X, et al. CRISPR/Cas9-mediated targeting of the Rosa26 locus produces Cre reporter rat strains for monitoring Cre/loxP-mediated lineage tracing. Febs J. 2017;284:3262.
Guan F, Yang X, Li J, Dong W, Zhang X, Liu N, et al. New molecular mechanism underlying Myc‐mediated cytochrome P450 2E1 upregulation in apoptosis and energy metabolism in the myocardium. J Am Heart Assoc. 2019;8:e009871.
Lu D, Ma Y, Zhang W, Bao D, Dong W, Lian H, et al. Knockdown of cytochrome P450 2E1 inhibits oxidative stress and apoptosis in the cTnTR141W dilated cardiomyopathy transgenic mice. Hypertension. 2012;60:81–89.
Michele U, Alex D, Martin F, Mervyn S. The key role of nitric oxide in hypoxia: hypoxic vasodilation and energy supply-demand matching. Antioxid Redox Signal. 2013;19:1690–710.
Lu D, Wang J, Li J, Guan F, Zhang X, Dong W, et al. Meox1 accelerates myocardial hypertrophic decompensation through Gata4. Cardiovasc Res. 2017;114:300–11.
Zierhut W, Studer R, Laurent D, KäStner S, Allegrini P, Whitebread S, et al. Left ventricular wall stress and sarcoplasmic reticulum Ca(2+)-ATPase gene expression in renal hypertensive rats: dose-dependent effects of ACE inhibition and AT1-receptor blockade. Cardiovasc Res. 1996;31:758–68.
Su J, Fang M, Tian B, Luo J, Jin C, Wang X, et al. Hypoxia induces hypomethylation of the HMGB1 promoter via the MAPK/DNMT1/HMGB1 pathway in cardiac progenitor cells. Acta Biochim Biophys Sin. 2018;50:1121–30.
Pian L, Li W, Xiao J, Cui J, Cohen P, Yu D, et al. Mitochondrial DNA hypomethylation is a biomarker associated with induced senescence in human fetal heart mesenchymal stem cells. Stem Cells Int. 2017;2017:1–12. 2017,(2017-4-6)
Siegfried Z, Eden S, Mendelsohn M, Feng X, Tsuberi BZ, Cedar H. DNA methylation represses transcription in vivo. Nat Genet. 1999;22:203–6.
Yuko T, Hideo E, Masaki O, En L, Hiromitsu N. De novo DNA methyltransferase is essential for self-renewal, but not for differentiation, in hematopoietic stem cells. J Exp Med. 2007;204:715–22.
Vujic A, Robinson EL, Ito M, Haider S, Ackers-Johnson M, See K. et al. Experimental heart failure modelled by the cardiomyocyte-specific loss of an epigenome modifier, DNMT3B. J Mol Cell Cardiol. 2015;82:174–83.
Ponikowski P, Anker SD, Chua TP, Szelemej R, Piepoli M, Adamopoulos S, et al. Depressed heart rate variability as an independent predictor of death in chronic congestive heart failure secondary to ischemic or idiopathic dilated cardiomyopathy. Am J Cardiol. 1997;79:1645–50.
Shehab A, Elnour AA, Struthers AD. Heart rate variability as an indicator of left ventricular systolic dysfunction. Cardiovasc J Afr. 2009;20:278–83.
Wei S, Yanyan Q, Huijun W, Xiaojing M, Ping Z, Long C, et al. Association between mRNA levels of DNMT1, DNMT3A, DNMT3B, MBD2 and LINE-1 methylation status in infants with tetralogy of Fallot. Int J Mol Med. 2013;32:694–702.
Arakawa Y, Watanabe M, Inoue N, Sarumaru M, Hidaka Y, Iwatani Y. Association of polymorphisms in DNMT1, DNMT3A, DNMT3B, MTHFR and MTRR genes with global DNA methylation levels and prognosis of autoimmune thyroid disease. Clin. Exp. Immunol. 2012;170:194–201.
Tao H, Yang JJ, Chen ZW, Xu SS, Zhou X, Zhan HY, et al. DNMT3A silencing RASSF1A promotes cardiac fibrosis through upregulation of ERK1/2. Toxicology. 2014;323:42–50.
Xuemei Q, Lihua Z, Sen L, Yunwei S, Yingbin L, Jiaojiao H, et al. Upregulation of DNMT1 mediated by HBx suppresses RASSF1A expression independent of DNA methylation. Oncol Rep. 2014;31:202–8.
Mitchell KA, Easwaran H, Baylin SB. Abstract 4779: DNMT3B (a de novo DNA methyltransferase) epigenetically regulates gene expression, independent of its DNA methyltransferase activity. Cancer Res. 2014;74:4779–4779.
Yi Q, Xu H, Yang K, Wang Y, Tan B, Tian J, et al. Islet-1 induces the differentiation of mesenchymal stem cells into cardiomyocyte-like cells through the regulation of Gcn5 and DNMT-1. Mol Med Rep. 2017;15:2511–20.
Watson CJ, Patrick C, Isaac T, Roisin N, Watson JA, Claire R, et al. Hypoxia-induced epigenetic modifications are associated with cardiac tissue fibrosis and the development of a myofibroblast-like phenotype. Hum Mol Genet. 2014;23:2176–88.
Felisbino MB, McKinsey TA. Epigenetics in cardiac fibrosis: emphasis on inflammation and fibroblast activation. JACC Basic Transl Sci. 2018;3:704–15.
Esteban OPE, Silvia MG, Patel JV, Mariano V, Francisco M, Lip GYH, et al. Role of microRNAs in cardiac remodelling: new insights and future perspectives. Int. J. Cardiol. 2013;167:1651–9.
Kao YH, Lien GS, Chao TF, Chen YJ. DNA methylation inhibition: a novel therapeutic strategy for heart failure. Int. J. Cardiol. 2014;176:232–3.
Funding
This research was supported by Chinese Academy of Medical Sciences (CAMS) Innovation Fund for Medical Sciences (CAMS-I2M, 2016-I2M-1-015), and National Natural Science Foundation (31872314).
Author information
Authors and Affiliations
Contributions
DL and L-FZ conceived and designed experiments; T-TW performed experiments and wrote the main manuscript text; Y-WM design the identification methods of animals; WD analyzed the echocardiographic data; XZ and SG performed the breeding and genotyping of the genetically modified animals; J-ZW performed the expression analysis in heart tissues.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Wu, TT., Ma, YW., Zhang, X. et al. Myocardial tissue-specific Dnmt1 knockout in rats protects against pathological injury induced by Adriamycin. Lab Invest 100, 974–985 (2020). https://doi.org/10.1038/s41374-020-0402-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41374-020-0402-y
This article is cited by
-
Hallmarks of cardiovascular ageing
Nature Reviews Cardiology (2023)
-
Integration of epigenetic regulatory mechanisms in heart failure
Basic Research in Cardiology (2023)
-
DNA methyltransferase 1 (DNMT1) suppresses mitophagy and aggravates heart failure via the microRNA-152-3p/ETS1/RhoH axis
Laboratory Investigation (2022)
-
Rat models of human diseases and related phenotypes: a systematic inventory of the causative genes
Journal of Biomedical Science (2020)
