
Abstract
Alzheimer’s disease is highly heritable and characterized by amyloid plaques and tau tangles in the brain. The aim of this study was to investigate the association between genetic predisposition, Aβ misfolding in blood plasma, a unique marker of Alzheimer associated neuropathological changes, and Alzheimer’s disease occurrence within 14 years. Within a German community-based cohort, two polygenic risk scores (clinical Alzheimer’s disease and Aβ42 based) were calculated, APOE genotype was determined, and Aβ misfolding in blood plasma was measured by immuno-infrared sensor in 59 participants diagnosed with Alzheimer’s disease during 14 years of follow-up and 581 participants without dementia diagnosis. Associations between each genetic marker and Aβ misfolding were assessed through logistic regression and the ability of each genetic marker and Aβ misfolding to predict Alzheimer’s disease was determined. The Alzheimer’s disease polygenic risk score and APOE ε4 presence were associated to Aβ misfolding (odds ratio, 95% confidence interval: per standard deviation increase of score: 1.25, 1.03–1.51; APOE ε4 presence: 1.61, 1.04–2.49). No association was evident for the Aβ polygenic risk score. All genetic markers were predictive of Alzheimer’s disease diagnosis albeit much less so than Aβ misfolding (areas under the curve: Aβ polygenic risk score: 0.55; AD polygenic risk score: 0.59; APOE ε4: 0.63; Aβ misfolding: 0.84). Clinical Alzheimer’s genetic risk was associated to early pathological changes (Aβ misfolding) measured in blood, however, predicted Alzheimer’s disease less accurately than Aβ misfolding itself. Genetic predisposition may provide information regarding disease initiation, while Aβ misfolding could be important in clinical risk prediction.
Similar content being viewed by others

Background
Alzheimer’s disease (AD) is a heritable neurodegenerative disease with pathological changes possible 15–20 years before symptoms1,2. The disease is characterized by amyloid plaques and tau tangles in the brain, which can be confirmed in vivo through biomarkers or definitively through postmortem examination3.
Cerebrospinal fluid (CSF)-derived or positron emission tomography (PET) imaging biomarkers have been widely established to detect neuropathological changes associated with AD even years before clinical symptoms are present4. Recently, amyloid beta (Aβ) has also been measured in blood, as a cost-effective and minimally invasive AD marker5.
The heritability of AD has been estimated as high as 79%2. Other than APOE ε4 (APOE4), many common variants with low effect sizes have been confirmed to play a role in AD genetic risk6. In very large genome-wide association meta-analyses, more than 20 risk loci have been confirmed6. Polygenic risk scores (PRS), which summarize this risk through the summation of risk variants weighted by effects, have been developed to characterize AD genetic risk7. AD PRSs have exhibited significant predictive ability of AD diagnosis7, however, significant associations with CSF or PET measured Aβ have been less consistent8,9,10,11. Although several genome-wide association studies have identified associated loci with abnormal Aβ levels measured in CSF or by PET imaging12,13,14,15,16, Aβ PRSs that may characterize Aβ specific risk have not been explored. Additionally, the relationship between AD/Aβ genetic risk, as distinguished through a PRS, and Aβ measured in blood has yet to be investigated.
The measurement of Aβ misfolding in blood is one strategy to identify early pathological changes associated to AD. In early stages of amyloid accumulation Aβ experiences a structural change from monomeric, alpha-helical or disordered conformations to β‐sheet‐enriched isoforms, the basis of plaque formation in the brain17,18,19.
The aim of this study was to investigate the association between various genetic predictors (APOE, AD PRS, and Aβ PRS), Aβ misfolding in blood plasma, a unique marker of Alzheimer associated neuropathological changes, and Alzheimer’s disease occurrence within 14 years. Additionally, the ability of the genetic risk markers and Aβ misfolding to predict vascular dementia (VD) diagnoses within 14 years was assessed.
Materials and methods
Study design and participants
The AD PRS was derived from results of stage 1 of the IGAP meta-analysis6, while the Aβ PRS was derived from a genome-wide association study (GWAS)12. Both PRSs were applied in a subsample within the prospective community-based cohort, the ESTHER study.
Summary statistics from stage 1 of the IGAP meta-analyses from Kunkle et al.6 were utilized, in which genotyped and imputed data on 11,480,632 SNPs was used to meta-analyze four previously-published GWAS consortia datasets consisting of 21,982 AD cases and 41,944 controls6.
The Aβ PRS was drawn from summary statistics of the Aβ GWAS by Deming et al. A GWAS of Aβ42, tau, and phosphorylated tau levels in CSF from 3146 participants across nine studies was completed to identify novel biological AD associated variants12. The associations between 7,358,575 SNPs and low Aβ42 measured in CSF were assessed.
The subjects for the analyses for this study were drawn from the ESTHER study, a large community-based cohort study conducted in Saarland, Germany20,21. A total of 9940 participants aged 50–75 years were recruited by their general practitioners (GPs) during a general health examination in a statewide study in Saarland, Germany in 2000–2002. Participants completed standardized self‐administered health questionnaires and provided blood samples. Information regarding age, sex, education, medical history, and lifestyle factors was collected at baseline through participant questionnaires and medical records. Follow‐up questionnaires, medical records, and biological samples were collected after 2, 5, 8, 11, 14, and 17 years. The ESTHER study was approved by the Ethics Committee of the Medical Faculty of Heidelberg University and of the Physicians’ Board of Saarland, and all participants gave written informed consent.
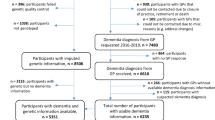
The subsample used for these analyses was a nested case-control study including 970 participants within the ESTHER study18. GPs reported patient dementia diagnoses and provided all available medical records from other specialized providers. The current guidelines in Germany for AD diagnosis follow the National Institute on Aging and the Alzheimer’s Association22 or the International Working group (IWG)-2 criteria23,24, for VD diagnosis the National Institute of Neurological Disorders and Stroke (NINDS)-Association Internationale pour la Recherche et l’Enseignement en Neurosciences (AIREN) criteria25. Excluded participants in this study included 184 participants without available genotyped or imputed genetic data for the PRSs, 15 cases where suspected dementia diagnosis could not be confirmed by further medical records, seven purported controls with a later identified dementia diagnosis, 34 participants without APOE genotype data, and one participant that withdrew informed consent (Fig. 1).

Flow chart of ESTHER study participants included in analyses.
Genotyping
Blood samples taken at baseline and stored at −80 °C were used for genetic analyses. DNA from whole blood samples was extracted using a salting out procedure. APOE data was directly genotyped and determined based on allelic combinations of the SNPs rs7412 and rs429358 using predesigned TaqMan SNP genotyping assays (Applied Biosystems, Foster City, CA). Genotypes were analyzed in an endpoint allelic discrimination read using the Bio‐RAD CFX Connect System (Bio‐Rad Laboratories, Hercules, CA).
Later, genome-wide SNP analyses were performed from extracted whole blood DNA using the Illumina Infinium OncoArray and Global Screening Array BeadChips (Illumina, San Diego, CA, USA)26. General genotyping quality control assessment was done following the Nature Protocols article from Anderson et al.27. Imputation of the quality controlled data was conducted using the Michigan Imputation Server, where SHAPEIT2 was used to phase the data and Minimac 4 was used to impute to the HRC Version r1.1 24 reference panel28,29.
Aβ misfolding measurement
The blood plasma samples used in this study were collected at baseline and used to measure Aβ misfolding in each participant. The Aβ misfolding marker assessed in these analyses detects the change in the Aβ peptides secondary structure through an immuno-infrared-sensor (WO 2015121339 A1), the details of which have been reported elsewhere18,30,31. This structure-based biomarker examines the misfolding state of Aβ in blood plasma. In agreement with a previously validated spectral threshold18, participants with a cutoff of < 1642 cm−1 were considered Aβ misfolding positive or to have high misfolding. Those participants at or above the validated spectral threshold were considered Aβ misfolding negative or to have low misfolding. The validated spectral threshold portrays the point at which the maximum position of the amide I absorbance band indicates an abnormal Aβ state associated to AD pathology. A plot of the distribution of Aβ misfolding can be found in Supplementary Fig. 1. At this threshold, we have previously shown 71 and 69% sensitivity and 91 and 86% specificity to detect AD cases in the ESTHER and BioFINDER studies, respectively18. Additionally, Aβ misfolding has shown significant correlations to Aβ burden measured by PET scan and in CSF18. The immuno-infrared-sensor has been validated in detail, including generation and characterization of NHS-silane, antibody batch-to-batch variation, antibody performance with synthetic and standard reference CSF and blood plasma samples, matrix effects, lower and higher limits of quantification, assay selectivity, sample handling, and documentation of zero background signals after Aβ immunodepletion17,18,30,31,32. The Aβ secondary structure distribution is detected as a relative measure and is independent of concentration fluctuations and sample variation. All plasma sample analyses were performed in a blinded manner at the department of Biophysics at Bochum University, Germany.
Polygenic risk score calculation
The AD PRS and Aβ PRS in this study were weighted scores including AD and Aβ associated SNPs, calculated by summing the number of risk alleles weighted by the magnitude of association to AD (ln of the odds ratio (OR)) from Kunkle et al.6 or Aβ from Deming et al.12.
For the AD PRS, SNPs reaching genome-wide significance in the IGAP meta-analysis were extracted from the imputed ESTHER data (n = 1234). Linkage disequilibrium-based clumping was carried out, providing the most significantly associated SNP in each region of linkage disequilibrium (using PLINK clumping command with a pairwise r2 threshold of 0.2) leaving 106 SNPs. Then, SNPs within or directly upstream/downstream from the APOE locus (chr19: 45,404,000–45,418,000) were excluded (n = 9). Finally, a minor allele frequency (MAF) threshold of 0.01 was applied excluding 25 SNPs. The remaining SNPs (n = 72) had imputation quality median R2 = 0.92 (R2 range: 0.47–0.99).
The same procedure was used for the Aβ PRS using summary statistics from Deming et al., where 133 SNPs were extracted from the imputed ESTHER data, and 21 SNPs remained after linkage disequilibrium-based clumping, all of which remained after applying the MAF threshold. The SNPs included in the Aβ PRS had imputation quality median R2 = 0.97 (R2 range: 0.83–0.99). Neither PRS included SNPs located within the APOE locus.
The scores were normalized by subtracting the mean and dividing by the standard deviation (SD), which were both calculated from the overall sample. For the sake of comparability of prediction performance of PRS and APOE, the cutoff for PRS+ was determined as the score point in which the number of PRS+ individuals was proportionate to the number of APOE4+ (≥ 1 ε4 allele) individuals in the Aβ misfolding negative group. It should be noted that this not a true or validated threshold but was chosen for comparability with APOE only. The distribution and cutoff values for each of the PRSs are described in Supplementary Fig. 1. Using the method described above, participants at or above the 73.6 percentile were considered AD PRS+ and those below AD PRS−. For the Aβ PRS, participants at or above the 74.7 percentile were considered Aβ PRS+ and those below Aβ PRS−. The PRSs were additionally investigated as continuous variables (per SD increase) and as categorical variables (quintiles).
Statistical analyses
Descriptive statistics were calculated to provide information on participant characteristics, while chi-square and t-tests were used to compare Aβ misfolding positive and Aβ misfolding negative groups in all participants and participants without dementia diagnosis.
Multivariable logistic regression models were used to assess the associations between the Aβ PRS, AD PRS, APOE, and Aβ misfolding, in cross-sectional analyses. Logistic regression was also used to assess the ability of Aβ misfolding (for comparison), both PRSs, and APOE4 to predict AD and VD diagnoses within 14 years. The PRSs were considered per SD increase in score, as a binary variable following the cutoff previously described, and as quintiles. APOE status was utilized as a binary variable (APOE4+:≥1 ε4 allele vs. APOE4−: no ε4 allele). Additionally, each of the PRSs and APOE4 status were combined and odds ratios were calculated for individuals that were: (1) both PRS+ and APOE4+; (2) PRS− and APOE4+; and (3) PRS+ and APOE4− all compared to the reference PRS− and APOE4−. Both Aβ PRS and AD PRS status were combined in a similar fashion. Covariates for all logistic regression analyses included age, sex, 10 principal components, and education, measured by years of formal education (≤9, 10–11, ≥12 years; standard categories of the German school system; the lowest category corresponds to a leaving certificate from school, the highest category corresponds to qualification for university). ORs including 95% confidence intervals (CIs) were calculated to assess associations with Aβ misfolding in all participants and in those participants without known dementia diagnosis. In the analyses with endpoints AD and VD, all participants were considered. Education data missing at random was imputed three times following the Markov chain Monte Carlo (MCMC) method33.
Receiver operating characteristic (ROC) curve analysis was completed for each PRS, APOE, and Aβ misfolding, where the PRSs and Aβ misfolding were considered continuously and APOE was considered categorically (APOE ε2ε2, ε2ε3, ε3ε4, ε4ε4 vs. ε3ε3). For AD diagnosis within 14 years, ROC curves and corresponding area under the curves (AUCs) were calculated based upon: (1) Aβ PRS; (2) AD PRS; (3) APOE; and (4) Aβ misfolding. Additionally, for explicit comparison with APOE, ROC curves were also calculated for (1) APOE + Aβ PRS; (2) APOE + AD PRS; (3) APOE + Aβ misfolding; and (4) for all predictors together APOE + AD PRS + Aβ PRS + Aβ misfolding. ROC contrast analysis using the DeLong test was conducted to compare for significant differences between curves34.
Additionally, AUC values were calculated for outcome Aβ misfolding (continuous) by the Aβ PRS (continuous), AD PRS (continuous), and APOE (categorical). Spearman rank correlation coefficients were calculated to assess the correlation between each of the genetic predictors (APOE: ordinal, i.e., number of APOE4 alleles, Aβ PRS: continuous, AD PRS: continuous).
All analyses were two-sided, conducted at an α-level 0.05, and completed using SAS software, version 9.4 (SAS Institute, Cary, NC).
Results
Participant characteristics
A flow chart of the participants included in this study is expressed in Fig. 1 and detailed participant characteristics are shown in Table 1. The analyses consisted of 728 participants with 114 participants considered Aβ misfolding positive and 614 participants Aβ misfolding negative. Of the 728 total participants, 59 had an AD diagnosis, 54 a VD diagnosis, 34 a mixed dementia diagnosis, and 581 remained without a dementia diagnosis throughout the 14-year follow-up.
The mean age of all participants was 68.5 years at ESTHER baseline when the blood samples were taken and used for Aβ measurements. Among Aβ misfolding positive participants, 27% were Aβ PRS+, 33% were AD PRS+, and 35% were APOE4+. Among Aβ misfolding negative participants, 25% were Aβ PRS+, 25% were AD PRS+, and 25% were APOE4+. This was due to the way in which the PRS cutoffs were selected (PRS positivity was determined as the score point in which the number of PRS+ individuals was proportionate to the number of APOE4+ (≥1 ε4 allele) individuals in the Aβ misfolding negative group).
Association of AD & Aβ genetic risk and Aβ misfolding
Among all participants
The AD PRS per SD increase in score and as a binary variable were significantly associated with Aβ misfolding (OR, 95% CI: AD PRS per SD: 1.25, 1.03–1.51; AD PRS+: 1.58, 1.01–2.46) (Table 2). APOE4 positivity was also significantly associated with Aβ misfolding (OR, 95%CI: 1.61, 1.04–2.49). The results of the PRSs as categorical variables (quintiles) can be found in Supplementary Table 1.
Among participants without dementia diagnosis
There were no significant associations between Aβ misfolding and any of the included predictors (Table 2).
Prediction of AD and VD diagnoses by Aβ and AD genetic risk
The ability of the Aβ PRS and AD PRS to predict AD diagnosis is shown in Table 3 and Supplementary Table 2. The prediction ability measured by AUC of the Aβ PRS, AD PRS, APOE, and Aβ misfolding as well as ROC contrast analyses for comparison of APOE to the additional predictors is depicted in Fig. 2.

A Area under the curve (AUC) values with 95% confidence intervals (CIs); B receiver operating characteristic (ROC) curves for individual predictors; C ROC curves for comparison of each predictor to APOE. AD, Alzheimer’s disease; APOE, apolipoprotein E; PRS, polygenic risk score. B AUC, 95% CI: Aβ PRS: 0.55, 0.46–0.63; AD PRS: 0.59, 0.51–0.68; APOE: 0.63, 0.55–0.70; Aβ misfolding: 0.84, 0.78–0.90C) AUC, 95% CI: APOE: 0.63, 0.55–0.70; APOE + AD PRS: 0.62, 0.53–0.70; APOE + Aβ PRS: 0.61, 0.53–0.69; APOE + Aβ misfolding: 0.86, 0.80–0.91; APOE + AD PRS + Aβ PRS + Aβ misfolding: 0.84, 0.78–0.91. P-values for ROC contrast analysis between APOE alone and APOE + additional predictor: APOE - (APOE + AD PRS): p = .52; APOE - (APOE + Aβ PRS): p = .26; APOE - (APOE + Aβ misfolding): p < .0001; APOE− (APOE + AD PRS + Aβ PRS + Aβ misfolding): p < 0.0001.
The Aβ PRS was predictive of AD diagnosis per SD increase in score (OR, 95%CI: 1.32, 1.01–1.73) and Aβ PRS+ participants had 85% greater odds of AD diagnosis than Aβ PRS− participants (OR, 95%CI: 1.85, 1.05–3.28). The AD PRS was also predictive of AD diagnosis per SD increase in score (OR, 95%CI: 1.47, 1.16–1.87) and AD PRS+ participants had 2.3 fold odds of AD diagnosis (OR, 95%CI: 2.29, 1.30–4.02). APOE4+ participants also had increased odds of AD diagnosis (OR, 95%CI: 2.69, 1.54–4.72).
Aβ misfolding exhibited superior AD diagnosis prediction ability compared to the genetic markers (AUC, 95% CI: Aβ PRS: 0.55, 0.46–0.63 AD PRS: 0.59, 0.51–0.68; APOE ε4: 0.63, 0.55–0.70; Aβ misfolding: 0.84, 0.78–0.90) (Fig. 2).
The relationship between the genetic risk markers, Aβ misfolding, and AD diagnosis is portrayed in Fig. 3 as AUC values and Spearman correlation coefficients. The genetic risk markers were moderately correlated, with correlation coefficients ranging from 0.49 to 0.52. The prediction ability of the genetic risk markers was greater for AD diagnosis than Aβ misfolding status. Among the genetic risk markers, APOE predicted both AD and Aβ PRS status. However, the prediction ability of Aβ misfolding for AD exceeded the prediction ability of any of the genetic markers.

The prediction of Aβ misfolding by the genetic risk markers and AD diagnosis by the genetic risk markers and Aβ misfolding is expressed as AUC values and the relationship between genetic risk markers is expressed as Spearman correlation coefficients.
The Aβ PRS, AD PRS, and APOE4 were not significantly predictive of VD diagnosis (Table 3). ROC curve analysis also revealed a lack of or minimal predictive ability of the genetic risk markers and Aβ misfolding to predict VD (AUC, 95% CI: Aβ PRS: 0.52, 0.45–0.61; AD PRS: 0.56, 0.48–0.64; APOE: 0.50, 0.42–0.57; Aβ misfolding: 0.54, 0.46–0.63).
Discussion
This study provides a comprehensive assessment of the interrelationship of various genetic predictors (APOE, AD PRS, and Aβ PRS), Aβ misfolding in blood, and AD diagnosis. Aβ misfolding in blood was significantly associated to an AD PRS and APOE4 status in a community-based cohort of older adults. There was a lack of association between the Aβ PRS and Aβ misfolding; however, the Aβ PRS significantly predicted AD diagnosis within 14 years. Aβ misfolding, a marker of early AD pathological changes, was far more predictive of AD diagnosis than the genetic risk markers.
Although this work is unique in the investigation of AD polygenic risk and Aβ misfolding in blood, previous studies have examined the relationship between an AD PRS and Aβ measured in CSF, by PET imaging, or postmortem. No previous study has utilized an Aβ specific PRS. Previous AD PRSs that included APOE exhibited positive associations to CSF Aβ10,35,36 or post-mortem measured Aβ37. However, in studies utilizing AD PRSs that excluded APOE, the association to CSF, PET, and post-mortem measured Aβ was mixed8,9,11,38,39,40,41,42, aligning with our results. AD PRSs have shown consistent associations to clinical AD diagnosis7, while the same/similar scores were less consistently associated to Aβ9,40,41.
Our results lacked significant associations between the Aβ PRS and Aβ misfolding, possibly because the Aβ PRS was based on a GWAS from Deming et al., where SNPs were identified with association to CSF-measured Aβ42, whereas Aβ misfolding examines the secondary structure changes of Aβ17. Additionally, while the GWA meta-analyses for AD have been large including 94,437 clinical AD cases6,43, Aβ specific GWAS have been smaller including at most 3,146 individuals12,15.
APOE has been more consistently reported to be significantly associated to Aβ11,35,36,40. It has been theorized that APOE contributes to amyloid accumulation and the AD PRS (additional associated variants) to AD conversion40. Additional associated variants included in the PRS were associated to clinical AD after symptom manifestation and, therefore, it may be possible that these variants could be associated to other drivers of AD progression, while APOE plays an inherent role in the disease initiation process of AD.
Interestingly, the associations between APOE/AD PRS and Aβ misfolding were not evident in participants without dementia diagnoses, possibly because those at high genetic risk of AD experience Aβ misfolding earlier and, therefore, also a diagnosis earlier. Although genetics play an important role in the development of AD, many other factors are involved in the manifestation of clinical AD44. AD is inherently very complex; with many medical, lifestyle, and social risk factors that play a role in development.
Aβ misfolding expressed greater AD predictive ability than any of the genetic markers. We have previously shown the odds of AD diagnosis in participants with high Aβ misfolding are 23 times that of participants without Aβ misfolding19. Aβ misfolding is a marker of early phenotypic manifestation of AD and can occur many years before clinical symptoms. While genetic risk markers provide important information regarding the risk and pathogenesis of AD, risk prediction by Aβ misfolding is stronger.
Implications
Most treatments after major Aβ accumulation have failed in the prevention of AD progression even when Aβ itself has been reduced45. Therefore, those at highest risk might benefit most from preventative treatment before amyloid accumulation. While there may be little clinical applicability without any available effective prevention or disease-modifying therapies, polygenic risk could play an important role in AD preventative research. Although PRSs can provide risk information at any time throughout life, even early in life before any amyloid accumulation, the predictive values for AD risk are low. It, therefore, appears questionable to what extent testing and preventative measures should be used based on such limited predictive value.
Aβ misfolding was shown to have much stronger predictive value for AD within 14 years before AD diagnosis, in a time frame where Aβ accumulation in the brain may still be limited. Aβ misfolding measurements could be crucial in identifying those individuals who would benefit most from AD symptom preventative measures and disease progression modifying therapeutic treatment. Further research should examine how early in the pathogenic process Aβ misfolding could identify those at highest AD risk and the effectiveness of preventative measures employed at that time point.
Strengths and limitations
The strengths of this study include the large community-based cohort that has been followed for over 14 years with available genetic, dementia, and Aβ blood plasma information. This study comprehensively assessed genetic predisposition (AD PRS, Aβ PRS, and APOE), early pathological changes (Aβ misfolding), and late phenotypic manifestation (AD diagnosis) providing novel information to the AD literature. Additionally, Aβ misfolding in blood plasma presents a unique marker of secondary structure changes of Aβ in blood plasma and beginning pathological changes associated to AD. Finally, the AD PRS is based upon the most recent GWA data6 and the investigation of an Aβ PRS is novel.
Several limitations of the study include the basis of the Aβ PRS, which was CSF measured Aβ and may therefore be not directly comparable to Aβ misfolding. Additionally, the nested case-control cohort study utilized for these analyses was only a small sample of the complete ESTHER cohort study with a much higher percentage of dementia cases than the entire cohort; however, similar associations between the AD PRS and dementia were evident in the entire cohort46. Another limitation includes the possibility of dementia misdiagnosis/underdiagnosis. The dementia diagnoses made in the ESTHER study were clinical diagnoses reported heterogeneously by numerous practitioners, and may be inferior to diagnostic standards that can be achieved in highly specialized academic settings. This is however the nature of community-based cohort studies, which portray common practice in such a setting. Also, the relatively small sample size and low number of AD and VD cases limited the power of the study and the generalizability of the results is limited to individuals of European descent.
Conclusion
Alzheimer’s genetic risk, defined by an AD PRS and APOE4, was significantly associated with Aβ misfolding, an early blood marker of AD associated pathology, in a community-based cohort of older adults, albeit somewhat inconsistently. An association between an Aβ PRS and Aβ misfolding was not evident, however, the Aβ PRS was predictive of AD diagnosis within 14 years. Aβ misfolding was much more predictive of AD than any of the genetic risk markers, asserting itself as a viable AD risk marker. Further research should thoroughly evaluate and compare the potential of risk stratification by genetic risk markers and Aβ misfolding for more effective and cost-effective targeted measures of precision prevention and disease-modifying treatment.
References
Van Cauwenberghe, C., Van Broeckhoven, C. & Sleegers, K. The genetic landscape of Alzheimer disease: clinical implications and perspectives. Genet. Med. 18, 421–430 (2016).
Gatz, M. et al. Role of genes and environments for explaining Alzheimer disease. Arch. Gen. Psychiatry 63, 168–174 (2006).
Jack, C. R. et al. NIA-AA Research framework: toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 14, 535–562 (2018).
Jack, C. R. Jr. et al. Brain beta-amyloid measures and magnetic resonance imaging atrophy both predict time-to-progression from mild cognitive impairment to Alzheimer’s disease. Brain 133, 3336–3348 (2010).
Zetterberg, H. & Burnham, S. C. Blood-based molecular biomarkers for Alzheimer’s disease. Mol. Brain 12, 26 (2019).
Kunkle, B. W. et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat. Genet. 51, 414–430 (2019).
Stocker, H., Möllers, T., Perna, L. & Brenner, H. The genetic risk of Alzheimer’s disease beyond APOE ε4: systematic review of Alzheimer’s genetic risk scores. Transl. Psychiatry 8, 166 (2018).
Voyle, N. et al. Genetic risk as a marker of amyloid-β and tau burden in cerebrospinal fluid. J. Alzheimers Dis. 55, 1417–1427 (2017).
Mormino, E. C. et al. Polygenic risk of Alzheimer disease is associated with early- and late-life processes. Neurology 87, 481–488 (2016).
Schultz, S. A. et al. Cardiorespiratory fitness alters the influence of a polygenic risk score on biomarkers of AD. Neurology 88, 1650–1658 (2017).
Martiskainen, H. et al. Effects of Alzheimer’s disease-associated risk loci on cerebrospinal fluid biomarkers and disease progression: a polygenic risk score approach. J. Alzheimers Dis. 43, 565–573 (2015).
Deming, Y. et al. Genome-wide association study identifies four novel loci associated with Alzheimer’s endophenotypes and disease modifiers. Acta Neuropathol. 133, 839–856 (2017).
Cruchaga, C. et al. GWAS of cerebrospinal fluid tau levels identifies risk variants for Alzheimer’s disease. Neuron 78, 256–268 (2013).
Liu C. et al. Genome-wide association and mechanistic studies indicate that immune response contributes to Alzheimer’s disease development. Front. Genet. 9, 410 (2018).
Chung, J. et al. Genome-wide association study of Alzheimer’s disease endophenotypes at prediagnosis stages. Alzheimers Dement 14, 623–633 (2018).
Li, Q. S., Parrado, A. R., Samtani, M. N. & Narayan, V. A., Initiative AsDN. Variations in the fra10ac1 fragile site and 15q21 are associated with cerebrospinal fluid aβ1-42 level. PLoS ONE 10, e0134000 (2015).
Nabers, A., Hafermann, H., Wiltfang, J. & Gerwert, K. Aβ and tau structure-based biomarkers for a blood-and CSF-based two-step recruitment strategy to identify patients with dementia due to Alzheimer’s disease. Alzheimers Dement. 11, 257–263 (2019).
Nabers, A. et al. Amyloid blood biomarker detects Alzheimer’s disease. EMBO Mol. Med. 10, e8763 (2018).
Stocker, H. et al. Prediction of Alzheimer’s disease diagnosis within 14 years through Aβ misfolding in blood plasma compared to APOE4 status, and other risk factors. Alzheimers Dement. 16, 283–291 (2019).
Raum, E. et al. Changes of cardiovascular risk factors and their implications in subsequent birth cohorts of older adults in Germany: a life course approach. Eur. J. Prev. Cardiol. 14, 809–814 (2007).
Löw, M., Stegmaier, C., Ziegler, H., Rothenbacher, D. & Brenner, H. Epidemiological investigations of the chances of preventing, recognizing early and optimally treating chronic diseases in an elderly population (ESTHER study). Dtsch. Med. Wochenschr. 129, 2643–2647 (2004).
McKhann, G. M. et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 7, 263–269 (2011).
Dubois, B. et al. Advancing research diagnostic criteria for Alzheimer’s disease: the IWG-2 criteria. Lancet Neurol. 13, 614–629 (2014).
Deutsche Gesellschaft für Neurologie. S3-Leitlinie“Demenzen” (Springer-Verlag GmbH Germany, 2016).
Román, G. C. et al. Vascular dementia: diagnostic criteria for research studies: report of the NINDS‐AIREN International Workshop. Neurology 43, 250–250 (1993).
Gao, X., Thomsen, H., Zhang, Y., Breitling, L. P. & Brenner, H. The impact of methylation quantitative trait loci (mQTLs) on active smoking-related DNA methylation changes. Clin. Epigenetics 9, 87 (2017).
Anderson, C. A. et al. Data quality control in genetic case-control association studies. Nat. Protoc. 5, 1564–1573 (2010).
Das, S. et al. Next-generation genotype imputation service and methods. Nat. Genet. 48, 1284–1287 (2016).
McCarthy, S. et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat. Genet. 48, 1279–1283 (2016).
Nabers, A. et al. Amyloid-β-secondary structure distribution in cerebrospinal fluid and blood measured by an immuno-infrared-sensor: A biomarker candidate for Alzheimer’s disease. Anal. Chem. 88, 2755–2762 (2016).
Nabers, A. et al. An infrared sensor analysing label‐free the secondary structure of the Abeta peptide in presence of complex fluids. J. Biophotonics 9, 224–234 (2016).
Budde, B. et al. Reversible immuno-infrared-sensor for the detection of Alzheimer´s disease related biomarkers. ACS Sens. 4, 1851–1856 (2019).
Schafer J. L. Analysis of Incomplete Multivariate Data, 444 (Chapman and Hall/CRC, 1997).
DeLong E. R., DeLong D. M. & Clarke-Pearson D. L. Comparing the areas under two or more correlated receiver operating characteristic curves: a nonparametric approach. Biometrics 44, 837-845 (1988)
Darst, B. F. et al. Pathway-specific polygenic risk scores as predictors of amyloid-β deposition and cognitive function in a sample at increased risk for Alzheimer’s disease. J. Alzheimers Dis. 55, 473–484 (2017).
Sleegers, K. et al. A 22-single nucleotide polymorphism Alzheimer’s disease risk score correlates with family history, onset age, and cerebrospinal fluid Abeta42. Alzheimers Dement 11, 1452–1460 (2015).
Tasaki, S., Gaiteri, C., Mostafavi, S., De Jager, P. & Bennett, D. A. The molecular and neuropathological consequences of genetic risk for Alzheimer’s dementia. Front Neurosci. 12, 699 (2018).
Yu, L. et al. Association of cortical β-amyloid protein in the absence of insoluble deposits With Alzheimer disease. JAMA Neurol. 76, 818–826 (2019).
Tan, C. H. et al. Polygenic hazard score, amyloid deposition and Alzheimer’s neurodegeneration. Brain 142, 460–470 (2019).
Leonenko, G. et al. Genetic risk for Alzheimer’s disease is distinct from genetic risk for amyloid deposition. Ann. Clin. Transl. Neurol. 6, 456–465 (2019).
Desikan, R. S. et al. Genetic assessment of age-associated Alzheimer disease risk: development and validation of a polygenic hazard score. PLoS Med. 14, e1002258 (2017).
Ge, T. et al. Dissociable influences of APOE ε4 and polygenic risk of AD dementia on amyloid and cognition. Neurology 90, e1605–e1612 (2018).
Lambert, J.-C. et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 45, 1452–1458 (2013).
Livingston, G. et al. Dementia prevention, intervention, and care. Lancet 390, 2673–2734 (2017).
Henley, D. et al. Preliminary results of a trial of atabecestat in preclinical Alzheimer’s disease. N. Engl. J. Med. 380, 1483–1485 (2019).
Stocker H. et al. Prediction of clinical diagnosis of Alzheimer’s disease, vascular, mixed, and all-cause dementia by a polygenic risk score and APOE status in a community-based cohort prospectively followed over 17 years. Mol. Psychiatry https://doi.org/10.1038/s41380-020-0764-y (2020).
Acknowledgements
The ESTHER study was supported by grants from the Baden‐Württemberg Ministry of Science, Research and Arts, the German Federal Ministry of Education and Research, the German Federal Ministry of Family, Senior Citizens, Women and Youth, the Saarland Ministry of Social Affairs, Health, Women and Family, and the Network Aging Research at Heidelberg University. HS is a doctoral student supported by a scholarship awarded from the Klaus Tschira Foundation. We thank the International Genomics of Alzheimer’s Project (IGAP) for providing summary results data for these analyses. The investigators within IGAP contributed to the design and implementation of IGAP and/or provided data but did not participate in analysis or writing of this report. This work received no specific funding.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The immuno‐infrared‐sensor was applied for patent by K.G. and A.N. (WO 2015121339 A1).
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Stocker, H., Nabers, A., Perna, L. et al. Genetic predisposition, Aβ misfolding in blood plasma, and Alzheimer’s disease. Transl Psychiatry 11, 261 (2021). https://doi.org/10.1038/s41398-021-01380-0
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41398-021-01380-0
This article is cited by
-
Copper metabolism-related Genes in entorhinal cortex for Alzheimer's disease
Scientific Reports (2023)
