
Abstract and Introduction
Abstract
Dyskinesia hampers the quality of life for most Parkinson's disease patients following several years of therapy. However, the severity of L-Dopa-induced dyskinesia (LID) varies between patients, being quite tolerable in late-onset patients. Understanding the pathogenesis of LID has contributed to the development of a set of therapeutic strategies, including the choice, in early stages, of the least pulsatile regimen of dopamine-receptor activation. In cases where LIDs are already disabling, there is only a limited number of options: the optimization of ongoing DOPA-centered treatment, the utilization of glutamate antagonists and the exploration of the benefits of antipsychotic agents. More radical solutions are provided by deep brain stimulation in the subthalamic nucleus (or internal pallidus). This approach has proved efficacious in reducing LID, largely because it allows a reduction in dopaminergic daily doses. Stereotactic neurosurgery has fuelled several lines of investigation regarding the crosstalk between the basal ganglia and motor cortex. Here, we will present interesting evidence highlighting the potential for repetitive transcranial stimulation in reducing the occurrence of LID. The future may disclose important new avenues for the treatment of LIDs, given the current development of promising agents that might target different facets of dyskinesia, such as the impairment of striatal plasticity and non-Dopaminergic contributors such as adenosine, nitric oxide and the nucleotide cascade.
Introduction
The definition of dyskinesia as a motor complication in the course of Parkinson's disease (PD) includes a spectrum of involuntary movements schematically characterized by the prominence of dystonic or choreic features with a variable occurance following the intake of dopaminergic drugs.
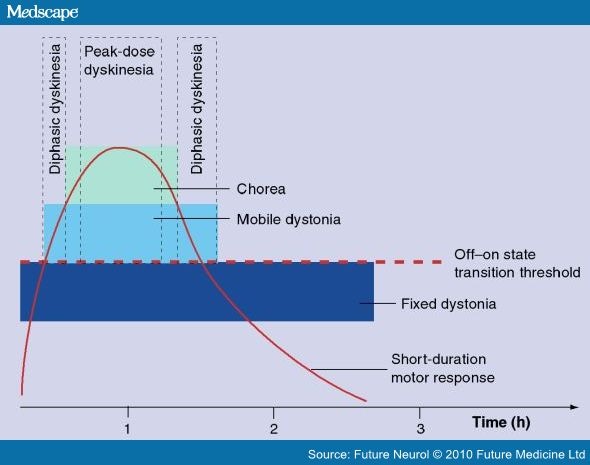
For the general practitioner, dyskinesias are considered only as the classical disturbance of the ON-period, the peak-dose L-Dopa-induced dyskinesias (LIDs). As a matter of fact, we should consider at least three categories of abnormal involuntary movements (AIMs); first, and the most common, is the 'peak-dose dyskinesia', which parallels L-Dopa concentration in the synaptic cleft (as does the short duration response [SDR]) (Figure 1); a second pattern of dyskinesia is the so-called 'biphasic' one, which is maximal at the beginning of the ON motor response, as well as at the end of the motor effect. A third pattern is defined as 'off-dystonia', which is usually painful, and manifests in the distal leg or foot.[1]

Figure 1.
The different dyskinesias present following an effective L-Dopa dose.
Reproduced with permission from [2].
The perception of dyskinesias may differ between neurologists and patients.[2] Many PD patients tend to underestimate AIMs, especially if not very severe, and find it difficult to indicate precisely, unless instructed to fill in diaries detailing to what extent their diurnal life is disturbed by disabling AIMs.
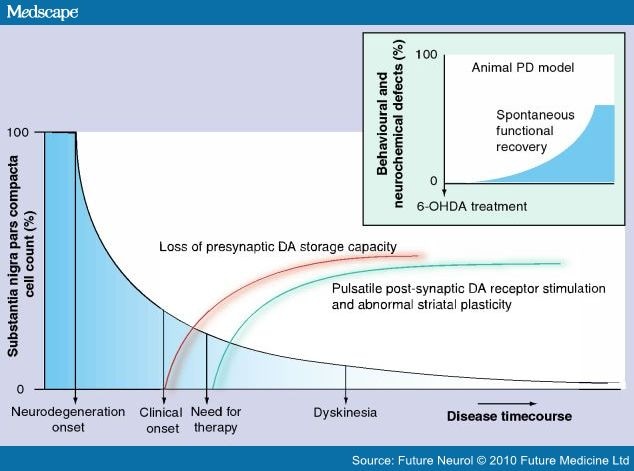
A deep analysis of dyskinesia pathogenesis is beyond the scope of this essay. A hypothetic cascade of events is summarized in Figure 2,[3–5] where we have tried to schematically highlight the pathogenetic factors, which have been inferred from Parkinsonian mammalian models but have also received some solid confirmation in human studies, through imaging or extracellular recordings during sterotactic neurosurgery. Of note is the distinction between 'induction processes'[6–10] and 'expression factors'.[11–17] Since our goal is mainly to examine current treatments for LID in PD patients (and not to review the literature on mammalian models) we felt it improper to discuss all experimental investigations. Besides, the vast majority of brilliant research studies in 6-hydroxydopamine (6-OHDA) or MPTP-treated mammals tend to underestimate the fact that the models (at least in their more common utilization) promote an extensive and fast-occurring neuronal loss, which is substantially different to the progressively occurring neuronal death observed in PD.[18] Hence, several inferences derived from those models, especially when the scientist's ambition is to interpret the complex development of complications related to the evolution of the human idiopathic disease, have to be viewed with extreme caution and may not provide consistent avenues for practical therapies.

Figure 2.
Factors that can induce dyskinesia.
The more relevant factors that are responsible for the induction of dyskinesia, outlined as the extent of nigral cell loss and the combined interplay of impairment of storage machinery and damage from modality of drug administration. On the right, full dyskinesia expression seems to be related to variety of additional processes, here synthesized in terms of impaired striatal plasticity and abnormal circuitry. Note, in the inset box, the putative time course of neurochemical deficits in standard modeling (adapted from [18]). When lesioning ex abrupto induces a greater than 90% sudden neuronal loss of tyrosine hydroxylase immunolabled elements, it has to be presumed that compensatory mechanisms come into action immediately and try to alleviate PD signs, a condition, of course, opposite to the slowly progressive development of behavioural signs in humans. This simple consideration should suggest great caution in utilizing the routine lesioning models as a proper tool to interpret cascades of events that underlie LDopa-induced dyskinesia in humans.
6-OHDA: 6-hydroxydopamine; DA: Dopamine; PD: Parkinson's disease
More critical, for the sake of our discussion centered on available and/or potential antidyskinesia therapies, is to analyze, focusing on clinical studies, the actual incidence and impact of LID.
On one hand, no consistent descriptions of dyskinesias were recorded pre-L-Dopa, as historically stated by Hoehn and Yahr,[19] while it has been demonstrated that approximately 40% of PD patients develop AIMs after 5–6 years of L-Dopa treatment.[20] The obvious implication of this evidence is that dyskinesias are strictly linked to L-Dopa-based therapy. If this is the case, how and when would L-Dopa exert prodyskinetic effects?
It used to be believed that a 'priming effect' was required to induce the later occurrence of dyskinesia.[3] Priming was considered to be the complex process by which dopaminergic receptors become sensitized after the first dopaminergic challenge in such a way that each administration of therapy modifies the response to subsequent drug administrations. In other words, dyskinesia would not be detectable at the beginning of treatment or after the very first dose, but would develop following repeated L-Dopa administration. As a consequence, this concept, which interprets dogmatically the correlation of LID with prolonged L-Dopa therapy, is currently accepted and obeys a sort of exhaustive cause–effect relationship. However, since the pioneering era of L-Dopa, Hornykiewitz and Birkmayer,[21] as well as Cotzias and colleagues,[22] have dramatically illustrated that even first, but high, doses of L-Dopa can cause AIMs, if administered to extremely severe and advanced PD patients. Of note, experimental studies in PD models have also highlighted the possibility that AIMS may be elicited on day 1 of the induction protocol, either in MPTP-intoxicated marmosets[23] or 6-OHDA lesioned rats.[24] As emerging literature have recently demonstrated, the extent of nigral dopaminergic denervation, as well as the impairment of both nigrostriatal storage and release mechanisms, combined with a nonphysiological (pulsatile) dopaminergic stimulaton with L-Dopa treatment, seem to be decisive events leading to abnormal striatal plasticity and circuitry changes; and thus, the expression of LID.[3–5]
Nadjar and coauthors recently challenged the 'priming' dogma.[25] They provide strong arguments in favor of the inference that the first drug administration does not induce, but instead unmasks or exacerbates sensitization mechanisms (the first drug dose would highlight the multiple maladaptive mechanisms that the neurodegeneration has already fuelled).
Hence, although we are aware of the importance of choosing the appropriate first antiparkinsonian treatment in de novo PD patients, with the dopamine (DA) agonists less prone to cause the development of dyskinesia compared with L-Dopa, an excessive emphasis has been put, in terms of pathogenesis, on the therapeutic history of patients manifesting dyskinesias or not. The chronicity and pulsatility of the treatment do not, per se, lead to LIDs, but certainly exacerbate the likelihood of developing LID, decrease the dyskinesia threshold dose of the drug[26] and shift the dyskinesia dose–response curve to the left.[5]
In this context, more research should be devoted towards discriminating the induction factors (degree of denervation and genetic predisposition), unmasking the mechanisms (improperly pulsatile dopaminergic stimulation and unwanted compensatory mechanisms), and eventually towards designing early neurorestorative therapies intended to restore striatal dopaminergic innervation.[25]
Nowadays, PD patients who are forced to delay any exogenous dopaminergic stimulation for years are becoming a rarity (and illogical cruelty); from a different perspective, in fact, it seems appropriate to anticipate a rational therapy, not just for symptomatic reasons, but also to postpone or halt the compensatory unwanted mechanisms promoted by DA depletion from the very early stages. Therefore, although the rate and severity of dyskinesias are principally related to the pharmacological therapy (pulsatile administration of priming-inducing agents, either L-Dopa or short-acting dopaminergic agonists), dyskinesia occurrence and induction should not only be viewed as a side effect of L-Dopa therapy (i.e., LID appearance seems to be unrelenting whatever strategy is used),[25] but rather as an obliged complication, a component of the disease progression – correlated with DA depletion and impaired DA endogenous release, and only partially correlated with the regimen of exogenously administered L-Dopa.
Another important aspect of the issue is the incidence of dyskinesia in relation to disease history. Classical studies demonstrated that dyskinesia affects less than 15% of PD patients treated for 5 years,[27] more than 30% of PD patients treated for 6–9 years and over 80% of PD patients treated for more than 10 years.[28]
In support of the importance of the disease duration, the extension of the Comparison of the Agonist Pramipexole versus Levodopa on Motor Complications of Parkinson's Disease (CALM-PD) study group by 6 years may dampen some dogmatic views when it states that, "dopaminergic motor complications (comprising dyskinesias) were more common in the initial L-Dopa group (68.4%) than in the initial pramipexole group (50%), although disabling dyskinesias were uncommon in both groups".[29]
The actual extent to which dyskinesias affect the quality of life of PD patients in more advanced stages is still a matter of dispute. At first, the incidence of LIDs in late-onset PD is much lower then in early-onset PD,[30] moreover, Ahlskog, in a provocative essay, suggested that we reconsider how tolerable involuntary movements may be in late-onset disease patients.[31] In community-based studies (not trials), such as that reported by Van Gerpen and coauthors,[32] 10 years of L-Dopa treatment was associated with a 43% risk of requiring medication adjustment because of LIDs and then, even so, dyskinesia was quite responsive to available therapies (only 12% of dyskinesias in patients were not satisfactorly controlled by medication changes).
Together, these observations, if we step aside from contentious and pharmaceutical-oriented debates, lead to the following statements:
Dyskinesia may be unobstructive and quite tolerable in late-onset PD patients; by contrast, an early onset PD patient (especially if treated early with L-Dopa) carries a higher risk of dyskinesias even after a few years of pharmacological treatment.[25]
Although the optimization of therapy from the onset, including the delay of L-Dopa therapy in favor of DA agonists with prolonged T1/2, and MAO-B inhibitors with a possible disease-modifying effect, contributes towards masking and even postponing dyskinesias, dyskinesias are not fully abolished by a therapy excluding L-Dopa, as demonstrated by the finding that DA agonists may also lead to the onset of dyskinesias.[2,33]
Dyskinesias remain a disturbing complication in the majority of advanced PD patients, despite the best clinical treatment and the possible decrease of the progression of motor impairment in advancing diseases stages.[34] LIDs may still represent an important cause of disability and social distress, contributing to the risk of falls and to the requirement for caregivers, especially in cases hampered by extrastriatal pathologies (i.e., memory complaints, hallucinations and comorbidities).[34]
This article will try to summarize:
The state-of-art therapeutic approaches with available molecular agents, including optimization of L-Dopa and DA-agonist treatment strategies in order to minimize the shift towards nonpulsatile control of clinical signs; ongoing utilization of nondopaminergic drugs with consistent experimental evidence of potential antidyskinetic effects. We will also acknowledge some interesting lines of investigation for future strategies.
The therapeutic possibilities offered by non-chemical techniques, including neurosurgical approaches and neurophysiological tools.
Future Neurology. 2010;5(2):277-299. © 2010 Future Medicine Ltd.
Cite this: Therapy for Dyskinesias in Parkinson's Disease Patients - Medscape - Mar 01, 2010.








Comments