Laboratory Studies
Results of all routine laboratory tests are within the reference range in Charcot-Marie-Tooth disease (CMT). Specific genetic tests are available for some types of CMT. A study by Murphy et al demonstrates that a molecular diagnosis can currently be achieved in over 60% of patients with CMT. [65] A molecular diagnosis is much more likely in patients with CMT-1 rather than CMT-2. Four genes commonly available for testing, PMP22 ( CMT-1A-PMP22 gene duplication; HNPP-PMP22 gene deletion), GJB1 ( CMT-1X), MPZ ( CMT-1B ), and MFN2 ( CMT-2A) account for over 90% of all CMT molecular diagnoses.
It is possible to narrow down the most likely gene based on nerve conduction studies and family history. Flow charts have been published using nerve conduction velocities to direct genetic testing, usually with the aid of family history information. [66]
-
CMT-1A - Pulsed-field gel electrophoresis or a specialized fluorescent in situ hybridization (FISH) assay is the most reliable genetic test to detect PMP22 gene duplication in CMT-1A and PMP22 gene deletion in HNPP. [67] DNA-based testing for the PMP22 duplication (CMT-1A) is widely available and detects more than 98% of patients with CMT-1A (see following image). Point mutations in the PMP-22 gene cause fewer than 2% of cases of CMT-1A and are identified by this technique. Approximately 70-80% of cases of CMT-1 are designated as CMT-1A, caused by duplication of the PMP-22 gene (locus 17p11). [16, 68] See the image below.
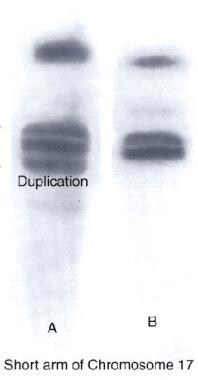 Charcot-Marie-Tooth disease type 1A DNA test showing duplication in the short arm of chromosome 17 (A) compared with normal (B).
Charcot-Marie-Tooth disease type 1A DNA test showing duplication in the short arm of chromosome 17 (A) compared with normal (B).
-
CMT-1C and CMT-1D - Very rarely, mutations occur in the EGR-2 (early growth response 2) gene or in the LITAF gene, causing CMT-1D and CMT-1C, respectively, for which molecular genetic testing also is clinically available.
-
CMT-2 - Clinically indistinguishable, the 4 subtypes of CMT-2 are distinguished solely from genetic linkage findings. The relative proportions of CMT-2A, CMT-2B, CMT-2C, and CMT-2D have not yet been determined. The chromosomal loci for CMT-2A, CMT-2B, CMT-2C, CMT-2D, CMT-2E, CMT-2F, CMT-2G, and CMT-2L have been mapped, but the genes have not been identified. Molecular genetic testing is clinically available only for CMT-2A, CMT-2B1, CMT-2E, and CMT-2F.
-
CMT-X - Molecular genetic testing of the GJB1 (Cx32) gene detects about 90% of cases. Such testing is clinically available.

Imaging Studies
High-resolution ultrasonography of the median nerve and other peripheral nerves may serve as an adjunct to electrodiagnosis in Charcot-Marie-Tooth disease type 1A. [5, 6, 7, 8, 9, 10]
Other Tests
Nerve biopsy rarely is indicated for the diagnosis of Charcot-Marie-Tooth disease (CMT), especially because genetic testing is available. Biopsies sometimes are performed in cases of diagnostic dilemmas. Findings vary in different types of CMT, as follows:
-
In CMT-1, peripheral nerves contain few myelinated fibers, and intramuscular nerves are surrounded by rich connective tissue and hyperplastic neurilemma. Lengths of myelin are atrophic along the fibers. Concentric hypertrophy of the lamellar sheaths is seen. Onion bulb formation, made up of circumferentially directed Schwann cells and their processes, frequently is observed.
-
In CMT-2, axon loss with wallerian degeneration generally is found.
-
In CMT-3, or Dejerine-Sottas disease, demyelination with thinning of the myelin sheath is observed.
-
Inflammatory infiltrate, indicating an autoimmune demyelinating process, should not be present.
Procedures
Electromyography/nerve conduction study (EMG/NCS) [5, 6, 7, 8, 9, 10]
-
If Charcot-Marie-Tooth disease (CMT) is suggested, perform an EMG/NCS first. Findings vary depending on the type of CMT.
-
In demyelinating types of CMT, such as CMT-1, diffuse and uniform slowing of nerve conduction velocities is observed (see following images).
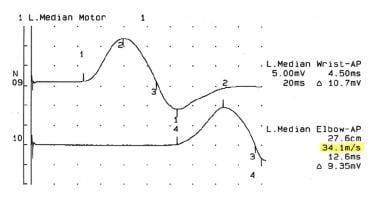 Nerve conduction study showing decreased nerve conduction velocity in the median nerve in an 18-year-old woman with Charcot-Marie-Tooth disease type 1.
Nerve conduction study showing decreased nerve conduction velocity in the median nerve in an 18-year-old woman with Charcot-Marie-Tooth disease type 1.
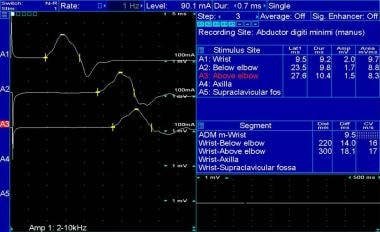 Right ulnar motor nerve conduction study in a 29-year-old patient with advanced Charcot-Marie-Tooth disease type 1.
Right ulnar motor nerve conduction study in a 29-year-old patient with advanced Charcot-Marie-Tooth disease type 1.
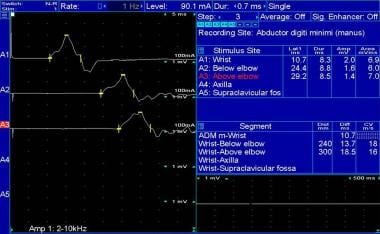 Left ulnar motor nerve conduction study in 29-year-old patient with advanced Charcot-Marie-Tooth disease type 1.
Left ulnar motor nerve conduction study in 29-year-old patient with advanced Charcot-Marie-Tooth disease type 1.
-
Harding and Thomas criteria for diagnosing CMT-1 include median motor nerve conduction velocity of less than 38 meters per second (m/s), with compound motor action potential (CMAP) and amplitude of at least 0.5 millivolts (mV). No focal conduction block or slowing should be present unless associated with other focal demyelinating processes.
-
All sensory and motor nerves that are tested show the same degree of marked slowing.
-
Absolute values vary, but they are approximately 20-25 m/s in CMT-1 and less than 10 in Dejerine-Sottas disease and congenital hypomyelination. Slowing of nerve conduction can also be found in asymptomatic individuals. In X- linked CMTs, motor nerve conduction velocities are faster than in CMT1A. [70]
-
In neuronal (ie, axonal) types of CMT, nerve conduction velocity usually is normal, but markedly low amplitudes are noted in sensory (ie, sensory nerve action potential [SNAP]) and motor (ie, CMAP) nerve studies.
-
In neuronal (ie, axonal) types of CMT, increased insertional activity is evident, with fibrillation potentials and positive sharp waves seen. Motor unit potentials show decreased recruitment patterns and neuropathic changes in morphology.



Histologic Findings
In Charcot-Marie-Tooth disease type 1 (CMT-1), peripheral nerves contain few myelinated fibers, intramuscular nerves are surrounded by a rich connective tissue and hyperplastic neurilemma, and lengths of myelin are atrophic along the fibers.
Concentric hypertrophy of the lamellar sheaths is seen. Formation of the typical onion bulb, made up of circumferentially directed Schwann cells and their processes, is noted.
In CMT-2, axonal degeneration is observed.
In CMT-3, Dejerine-Sottas disease, demyelination with thinning of the myelin sheath can be seen. [71]
No inflammatory infiltrate should be present, indicating an autoimmune demyelinating process.
-
Foot deformities in a 16-year-old boy with Charcot-Marie-Tooth disease type 1A.
-
Charcot-Marie-Tooth disease type 1A DNA test showing duplication in the short arm of chromosome 17 (A) compared with normal (B).
-
Nerve conduction study showing decreased nerve conduction velocity in the median nerve in an 18-year-old woman with Charcot-Marie-Tooth disease type 1.
-
Foot of 29-year-old with advanced Charcot-Marie-Tooth disease type 1.
-
Hands of 29-year-old with advanced Charcot-Marie-Tooth disease type 1.
-
Right ulnar motor nerve conduction study in a 29-year-old patient with advanced Charcot-Marie-Tooth disease type 1.
-
Left ulnar motor nerve conduction study in 29-year-old patient with advanced Charcot-Marie-Tooth disease type 1.
Tables
CMT Type |
Chromosome; Inheritance Pattern |
Age of Onset |
Clinical Features |
Average NCVs§ |
CMT-1A (PMP-22¶ dupl.) |
17p11.2; AD* |
First decade |
Distal weakness |
15-20 m/s |
CMT-1B (P0 -MPZ)** |
1q23.3; AD |
First decade |
Distal weakness |
< 20 m/s |
CMT-1C (non-A, non-B) (LITAF) |
16p13.13;AD |
Second decade |
Distal weakness |
26-42 m/s |
CMT-1D (EGR-2)# |
10q21.3; AD |
First decade |
Distal weakness |
15-20 m/s |
CMT-1E (PMP22) |
17p11.2; AD |
First decade |
Distal weakness, deafness |
15-20 m/s |
CMT-1F (NEFL) |
8p21.2; AD |
First decade |
Distal weakness |
15-20 m/s |
Xq13; XD‡ |
Second decade |
Distal weakness |
25-40 m/s |
|
CMT-2A |
1p36; AD |
10 y |
Distal weakness |
>38 m/s |
CMT-2B |
3q21; AD |
Second decade |
Distal weakness, sensory loss, skin ulcers |
Axon loss; Normal |
CMT-2C |
12q23-q24, AD |
First decade |
Vocal cord, diaphragm, and distal weakness |
>50 m/s |
CMT-2D |
7p14; AD |
16-30 y |
Distal weakness, upper limb predominantly |
Axon loss; N†† |
CMT-2E |
8p21; AD |
10-30 y |
Distal weakness, lower limb predominantly |
Axon loss; N |
CMT-2F |
7q11-q21; AD |
15-25 y |
Distal weakness |
Axon loss; N |
CMT-2G |
12q12-q13; AD |
9-76 y |
Distal weakness |
Axon loss; N |
CMT-2H |
8q21; AD |
15-25 y |
Distal weakness, pyramidal features |
Axon loss; N |
CMT-2I |
1q23; AD |
47-60 y |
Distal weakness |
Axon loss; N |
CMT-2J |
1q23; AD |
40-50 y |
Distal weakness, hearing loss |
Axon loss; N |
CMT-2K |
8q13-q21; AD |
< 4 y |
Distal weakness |
Axon loss; N |
CMT-2L |
12q24; AD |
15-25 y |
Distal weakness |
Axon loss; N |
CMT–R-Ax (Ouvrier) |
AR |
First decade |
Distal weakness |
Axon loss; N |
CMT–R-Ax (Moroccan) |
1q21; AR |
Second decade |
Distal weakness |
Axon loss; N |
Cowchock syndrome |
Xq24-q26 |
First decade |
Distal weakness, deafness, intellectual disability |
Axon loss; N |
HNPP|| (PMP-22 deletion) [55] or tomaculous neuropathy |
17p11; AD |
All ages |
Episodic weakness and numbness |
Conduction Blocks |
Dejerine-Sottas-syndrome (DSS) or HMSN-3 [56] |
P0; AR PMP-22; AD 8q23; AD |
2 y |
Severe weakness |
< 10 m/s |
Congenital hypomyelination (CH) |
P0, EGR-2 or PMP-22 AR |
Birth |
Severe weakness |
< 10 m/s |
CMT-4A [57] |
8q13; AR |
Childhood |
Distal weakness |
Slow |
CMT-4B (myotubularin- related |
11q23; AR |
2-4 y |
Distal and proximal weakness |
Slow |
CMT-4C |
5q23; AR |
5-15 y |
Delayed walking |
14-32 m/s |
CMT-4D (Lom) (N-myc downstream- regulated gene 1) |
8q24; AR |
1-10 y |
Distal muscle wasting, foot and hand deformities |
10-20 m/s |
CMT-4E (EGR-2) |
10q21; AR |
Birth |
Infant hypotonia |
9-20 m/s |
CMT-4G |
10q23.2; AR |
8-16 years |
Distal weakness |
9-20 m/s |
CMT-4H |
12p11.21-q13.11; AR |
0-2 years |
Delayed walking |
9-20 m/s |
CMT-4F |
19q13; AR |
1-3 y |
Motor delay |
Absent |
*Autosomal dominant †Autosomal recessive ‡X-linked dominant §Nerve conduction velocities ||Hereditary neuropathy with liability to pressure palsy ¶Peripheral myelin protein #Early growth response **Myelin protein zero ††Normal |
||||







