
Systemic RV Infection
The remainder of this review will deal with systemic aspects of RV infection. Data derived from humans and animal model systems will be considered. Evidence of infection at peripheral sites will be considered first, since these were the earliest data examined. Viremia and antigenemia will be considered second, since they have been demonstrated most recently.
Evidence for Systemic RV Infection
Evidence for Systemic Infection in Children. Work on human RVs dates from the demonstration, in 1973, that RV was present in the duodenum of children with acute diarrhea.[55] Since that time, sporadic clinical case reports described systemic disease that was temporally associated with RV infection and suggested spread of infection beyond the GI tract. The sporadic nature of these reports indicates that if systemic spread occurs it may be a rare event, or if spread is a common event, it rarely results in systemic disease. Indeed, it is controversial whether RV association with systemic disease is causative or if the virus is simply a passenger in a disease of different etiology. The recent demonstration of frequent viremia in children[56,57] suggests that systemic disease may be a rare outcome of spread. However, if systemic disease occurred in only one in 10,000 (0.01%) of 111 million RV-infected children each year, approximately 11,000 cases of systemic disease would be expected annually.
Within a decade of discovery, human RV was reported in association with systemic clinical symptoms. Early reports included association of RV infection with exanthem subitum,[58] CNS involvement,[59] intussusception,[60] elevated liver markers in serum[61] and fatalities of no other known etiology.[62] While the temporal association of these clinical symptoms with RV infection did not prove direct RV causation, studies that would tighten the RV systemic clinical syndrome link emerged in the next 20 years.
Liver involvement during RV infection was reported sporadically, generally manifested by abnormal liver function tests[63,64] or transient hepatitis.[65] Liver involvement was severe in immunocompromised children with chronic RV diarrhea, where RV antigen was detected in liver and kidney and active RV replication in the liver was documented.[66] Many reports of concomitant RV diarrhea and convulsions or seizures[67,68] suggested neurological involvement. Indeed, RV RNA was demonstrated in the cerebrospinal fluid (CSF) by reverse transcriptase PCR[69] and another study indicated that the presence of RV in the CSF did not originate as contamination of samples.[70] One study showed that RV RNA from the stool and CSF of an infected child had nearly identical VP4 and VP7 sequences and these sequences were nearly identical to the virus contemporaneously circulating in the community.[71] This shows a potential relationship between RV infection and CNS symptoms and the virus present in the community.
A number of reports associated RV infection with respiratory symptoms in children. An early prospective study of RV in infants and children found significantly more respiratory symptoms in children positive for RV than in children positive for diarrhea of other etiologies. Furthermore, respiratory symptoms appeared first in 15 out of 62 (24%) subjects who subsequently developed RV diarrhea.[72] In another study, tracheal aspirates were examined for RV and 16 out of 58 (27%) children with clinically diagnosed pneumonia had stool RV, although only six out of 16 ever had diarrhea.[73] Finally, infectious virus and RV-infected cells were detected in 25 out of 89 (28%) children sampled by oropharyngeal aspiration. The rate of RV at this site surpassed the combined rate of other viruses commonly found at this site.[74] Data such as these raised the question of whether RV can be transmitted by the respiratory route, a question that remains unresolved. Virus could also enter the respiratory tract upon vomiting, which is a common symptom of RV infection.
RV antigens were detected in myocardium from cases of unexpected death.[75] Pancreatitis, with transient islet cell antibodies, has been reported in association with an RV infection that also caused hypoglycemia-associated convulsions,[76] and RV has been shown to undergo complete replication cycles in primary nonhuman primate islet cells.[77] Another case report suggested the development of kidney stones secondary to RV dehydration resulting in sustained hypovolemia.[78] Finally, two out of three children who died of RV-associated disease were found to have RV antigen and RNA in a wide array of tissues, including spleen, heart, lung, kidney, testis, bladder, adrenal gland and pancreas.[79] The increasing reports of extraintestinal RV over time probably reflects the development of more sensitive methods to detect viral antigen and RNA. Table 1 summarizes the findings from selected publications that suggest RV is associated with systemic syndromes in humans.
Evidence for Systemic Infection in Animals. Diarrheal diseases have long been a chronic problem in the livestock industry, causing mortality, delay in weight gains and economic losses. In 1943, the first demonstration that a virus caused diarrhea came from feeding calves a cell-free filtrate of stool from a diarrheic child.[80] Subsequently in 1969, a virus was isolated from calves with diarrhea and characterized as 'reovirus like'.[81] Rapidly thereafter, similar reovirus-like viruses causing diarrhea were isolated from many wild and domestic mammalian species and from domestic fowl. These studies stimulated the search for human diarrhea viruses.
Work with the animal RVs concentrated on the diarrheal disease produced in neonates. The disease was severe. It was shown that antigenically different types existed and protection could be mediated by feeding colostrum near the time of infection. Passive vaccination strategies were developed that reduced morbidity in the field.[82]
The severe disease and fatalities in animals appear to result from the effects of diarrhea - dehydration with weight loss, susceptibility to other enteric pathogens and a weakened state leading to fatality resulting from severe changes in the environment. However, there were few reports of systemic disease that could not be assigned as secondary to diarrhea and dehydration.[82] The only evidence of systemic involvement in RV infection forthcoming from studies of animal disease was a single description of antigenemia in calves[83] and descriptions of viral antigen in systemic organs of colostrum-deprived pigs.[84,85] The only other description of systemic infection was the finding of infectious RV at systemic sites in the studies of EDIM virus.[50,51,52]
Studies on RV Spread to Systemic Tissues
Studies of Spread to Extraintestinal Tissues in Animal Models. Studies on the spread of RVs in humans are only possible at sites where samples can be collected by minimally invasive procedures, such as biopsies of the duodenum, blood and serum samples, and CSF. Thus, much of our understanding of RV spread has come from experiments performed in animal models. These studies are driven by the desire to determine if the extraintestinal spread of RV is clinically significant and the continued development of sensitive methods to detect RV or its components. Here, unless otherwise stated, all animals were inoculated orally, the natural route of RV infection. A concise summary of selected studies described is in Table 2 .
As noted previously, one of the first studies of RV found infectious virus in many tissues of infant mice.[50,51,52] Since blood was not removed from the tissues in these studies, the possibility remained that infectious virus in the tissues resulted from viremia and not infection at the peripheral site. However, early studies of the distribution of viral antigen outside the GI tract found antigens in the intestinal lymphoid tissues (Peyer's patches [PP] and mesenteric lymph node [MLN]), consistent with either antigen presentation for immune response or viral escape from the intestine.[86] Other early studies found RV antigen in the serum of experimentally infected calves[83] and in the lung, brain and spleen of colostrum-fed or -deprived experimentally infected pigs.[84,85] These studies suggested systemic involvement in large animal models, as well as in mice.
The report that RV caused chronic infection in severely immunocompromised children with virus shedding and diarrhea intermittently for up to 2 years[87] prompted the study of RV infection in immunocompromised mice. Murine RV caused persistent infection of CB-17 (severe combined immunodeficiency [SCID]) mice following oral infection, whereas normal mice cleared the infection in approximately 10 days.[88] Further studies using the rhesus RV (RRV) showed that 84% of SCID mice infected with RRV developed hepatitis, with a mortality rate of 27%. The surviving SCID animals developed a chronic liver disease with histologic lesions of diffuse hepatitis with focal parenchymal necrosis. Infectious RRV was detected in the liver in 100% of the SCID mice.[89] In immunocompetent BALB/c mice, hepatitis was noted in 27% of RRV infections with no mortality and symptoms resolved in 2-4 weeks. Infectious RRV was found in the liver in 85% of the RRV-infected BALB/c mice. Another virus strain, (WC3) did not escape the gut in either SCID or normal mice.[89] These studies demonstrated that RV escaped the intestine and caused a particularly severe hepatitis in immunodeficient mice; however, immunodeficiency was not required for escape from the gut as immunocompetent mice also had hepatitis, although at a reduced rate. Thus, RV could potentially escape the gut of orally infected mice and cause disease in the liver. The fact that RRV escaped, while WC3 did not, suggested that escape from the gut was determined by the virus.
The report that group C RV appeared to be associated with extrahepatic biliary atresia (EHBA) in children,[90] prompted a determination of the association of group A RV with EHBA since some cause hepatobiliary disease.[91] Two hepatobiliary-tropic RV strains (HCR-3, RRV) were inoculated orally into 2-day-old BALB/c mice. HCR-3 inoculation resulted in hepatobiliary disease in 24% of pups with 13% mortality and RRV resulted in 42% disease and 21% mortality. Approximately 75% of liver and biliary tract samples from HCR-3- and RRV-inoculated animals were positive for RV by culture or immunofluorescence (IF) 8 days postinoculation. Gross examination showed variable pathology and histopathology showed that inoculation with either HCR-3 or RRV was associated with an acute inflammation of the bile ducts with constriction soon after infection. In the liver parenchyma, inflammation was noted early, followed by a progression of changes, including focal necrosis, portal hepatitis, bile ductule proliferation and fibrosis.[91] The symptoms closely resemble EHBA in children, suggesting that the mouse may provide a tractable model for the study of EHBA and an association with group A RV.
Subsequently, a mouse model for EHBA using intraperitoneal inoculation of RV was developed. This route of infection bypasses gut-associated barriers to virus spread but results in a much higher frequency of biliary obstruction.[92] In this model, BALB/c mice have higher rates of cholestatic disease and lethality than other mouse strains. The disease was diverse but inflammatory and progressive, ultimately resulting in complete blockage of the bile duct.[92,93] The fraction of inoculated pups exhibiting EHBA increased as infection came closer to parturition, with 100% lethality attained when inoculation was within 12 h of birth.[94] Infection of pregnant dams or immunization of dams prevented the development of EHBA with the protection transferred transplacentally rather than lactogenically.[94] Treatment of infected pups with IFN-α daily for 7 days after onset of cholestatic disease allowed recovery in 76% of the pups.[95] In another study, RV infection of BALB/c mice resulted in 80% jaundice and 90% fatality due to obstruction of extrahepatic bile ducts.[96] Immunofluorescence revealed RV has a tropism for cholangiocytes as it colocalizes with cytokeratin-7, a marker for the epithelium of the extrahepatic bile duct. Infection of the duct epithelium triggers the infiltration of IFN-γ-secreting CD4+ and CD8+ lymphocytes. By contrast, IFN-γ-/- knockout mice, while still experiencing jaundice, did not develop the inflammatory obstruction of the extrahepatic bile ducts. Furthermore, daily administration of IFN-γ to the knockout mice for 14 days from the day of infection resulted in the restoration of extrahepatic biliary obstruction as in wild-type mice.[96] As was seen for viral escape to the liver, tropism of RV to the biliary tree is virus strain specific and it was suggested that tropism of RV to the biliary epithelium may be a property determined by the viral attachment protein VP4.[97] While the mouse model of biliary atresia is quite robust, the infectious etiology and RV involvement in biliary atresia of children remains controversial.[91,98]
The suggestion that extraintestinal spread of RV following oral inoculation was strain specific[89] led to the study of the viral genetic determinants of intestinal escape and extraintestinal infection. In this study, the virus stains RRV (hepatotropic), SA11-Cl4 (nonhepatotropic) and reassortants derived from RRV/SA11-Cl4 were used to orally infect 5-day-old strain CD-1 outbred mice.[53] Detection of infectious virus in the liver was used as a proxy for extraintesinal spread. Both RRV and SA11-Cl4 replicated in the intestinal tract, although with different kinetics; SA11-Cl4 was present early and disappeared, while RRV was present throughout. RRV/SA11-Cl4 reassortants segregated the kinetics of gut replication with RRV genome segment 7 (p < 0.00001), but other genes were also significant, although to a lesser degree (p = 0.01). In the reassortants, the presence of infectious virus in the liver also segregated with RRV genome segment 7 (p ~ 0.001) and no other segments were significantly biased in parental origin. The high significance of RRV genome segment 7 with titer in the gut suggested that escape of virus from the gut might correlate with a high titer in the gut. However, comparisons of gut titer in pups with virus in the liver and those without virus in the liver showed no significant linkage between gut titer and escape from the intestine. The high significance of RRV genome segment 7 with gut growth is probably an artifact of the late time when pups were sampled, so that reassortants with SA11-Cl4 segment 7 had reached peak titer and the titer had significantly decayed by the time of sampling. These data indicate that RRV genome segment 7 encodes the determinant of extraintestinal escape to the liver.[53] Segment 7 encodes viral NSP3, a protein known to function in the regulation of viral and host protein synthesis during infection[4] and possibly in RNA synthesis.[99] Sequence analysis of NSP3 from RRV and SA11-Cl4 revealed 82% amino acid identity and 91% similarity. The differences did not fall into any known functional domain of NSP3, so the mechanistic basis of NSP3 determination of extraintestinal escape phenotype remains unknown.[53] However, since NSP3 is a NSP, these data imply that active infection plays a role in escape.
RRV/SA11 reassortants were also used to examine the pathway of RV escape from the gut.[100] Following oral inoculation of 5-day-old strain CD-1 mice, RRV and a spread-competent reassortant (contains RRV genome segment 7) sequentially appeared in the gut, PP, MLN and liver, indicating that spread-competent viruses moved via a lymphatic route. By contrast SA11-Cl4 was not found outside the intestine. Surprisingly, the spread-incompetent reassortant (contains SA11-Cl4 segment 7) was found sequentially in the gut, PP and MLN but never in the liver, whereas it was not expected to spread. This suggested there were multiple determinants of spread from the gut to the MLN. To examine this, reassortants with SA11-Cl4 segment 7, while reassorting the remaining segments, were examined. In this subset of reassortants, the spread was as far as the MLN and the titer attained at that site segregated with RRV segment 6. Thus, reassortants with RRV segment 6 and SA11 segment 7 could escape the gut as far as the MLN (p = 0.001). However, only reassortants with RRV segment 7 could move to sites beyond the MLN (p = 0.001), regardless of the parental origin of the other genes. This study also implicates RRV NSP3 as the determinant of spread and suggests that the virus moves through the lymphatic system in an infected migratory cell.[100] The mechanism by which different genes function as determinants of spread to different points is not understood.
Rotashield, a live-reassortant vaccine with RRV as the backbone, was compared with RotaTeq, a live-reassortant vaccine with bovine WC3 as the backbone, in orally inoculated suckling CB17 (SCID) mice.[101] The RRV backbone of Rotashield was clearly hepatropic, whereas the WC3 backbone of RotaTeq appeared not to be hepatotropic, in SCID and normal animals.[89] After a total of 6 days postinoculation, 62% of Rotashield mice versus 0% of RotaTeq mice had hepatitis and/or bile duct obstruction. For Rotashield, only the RRV (G3) component of the vaccine was recovered from extraintestinal tissues, including spleen, MLN, liver, pancreas, blood cells and serum. By contrast, extraintestinal virus was rarely recovered from Rotateq-inoculated mice and was found in the liver of three mice at low titer, and in the pancreas and blood of one mouse each. The low-titer virus in the liver of RotaTeq-inoculated mice did not cause hepatic pathology. Since the components of Rotashield differ only in VP7, the authors speculated that genome segment 9, encoding VP7, may be critical in the hepatotropism of RRV,[101] a speculation that conflicts with the findings of others.[53,97,100] Whatever the basis of RRV hepatotropism, RRV appears to have been an unfortunate choice of genetic background for the Rotashield vaccine.
The studies discussed all relied on the detection of virus antigen and/or infectious virus at extraintestinal sites. Recently, a highly sensitive nucleic acid-based detection method, strand-specific quantitative reverse transcription PCR (ssQRT-PCR), was applied to study the extraintestinal replication of virus strains EC (mouse) or RRV (monkey) following the oral inoculation of 5-day-old BALB/c mice.[102] Control experiments showed that using ssQRT-PCR the presence of an approximately 1.5-fold excess sense stranded RNA over antisense stranded RNA was indicative of virus replication in a tissue, whereas more equal amounts of sense and antisense strands of RNA indicated nonreplicating virus was present. Additionally, this study was the first to allow direct comparison of the levels of virus and replication in various tissues of the same animal, thus providing information on whether virus in peripheral organs represented virus produced in the gut or resulted from replication in the peripheral tissue.
The conclusion that both EC and RRV have the ability to escape the small intestine, spread systemically and replicate in peripheral organs is based on the following results. EC and RRV viruses were found in the small intestine of 98 and 16% of the inoculated mice, respectively. EC replicated to more than 10,000-fold higher RNA copy number in the intestine than did RRV. RNA from both viruses was found for longer periods than fecal antigen detection shedding assays indicate and 86% of RRV mice shed virus even though RNA was in the small intestine of only 16%. EC (80%) and RRV (72%) viruses were both found in the MLN. The MLN was the extraintestinal site with the greatest replication for both viruses. Replication in the MLN occurred at similar levels for each virus, approximately 2 log10 above that in the blood. Furthermore, virus replication in the MLN persisted over the entire course of the experiment (15 days) and exceeded the time of replication in any tissue, including the small intestine. The strong replication in the MLN confirmed the results of others[53,100] and supports the notion that the MLN may be the portal for escape from the small intestine by providing a systemic site of prolonged replication. EC (64%) and RRV (10%) viruses were both also found in the liver.[102] EC replicated in the liver for a period of 6 days to levels exceeding those in the blood, whereas RRV replication was detected on only 2 days and the level of replication did not exceed the level of virus in the blood.[102] This finding is curious, since RRV is hepatopathogenic and a different murine strain (EDIM) was not hepatopathogenic.[89] Thus, it is unclear why RRV is more hepatopathogenic than murine RV, but the difference does not reflect an increased ability of RRV to replicate in the liver. Unfortunately, the biliary tree was not examined in these experiments so one does not know if damage to the bile duct is more common with RRV than with EC. In the absence of data, it is tempting to speculate that liver damage in RRV results from blockage of the bile duct leading to hepatocyte damage. EC (28%) and RRV (34%) viruses were found in the lung. EC appeared to replicate in the lung in a few animals, but RRV did not. However, RRV was found in the lung at levels exceeding those in the blood. Finally, EC (45%) virus was found in the kidney and replicated to levels above that in the blood for 6 days. By contrast, RRV (3%) virus was found only sporadically and no replication occurred in the kidney. In most cases in this study, replication in the liver as detected with ssQRT-PCR was confirmed by ELISA for viral antigen in the same tissue, or the demonstration of the presence of the viral replication by IF using a monoclonal antibody to the NSP NSP4.[102]
The association of RV infection with transient pancreatitis in children[76] and the ability of RV to replicate in primate islet cells,[77] led to a study of the effects of RV on diabetic mice.[103] When 5-day-old nonobese-diabetic (NOD) mice were orally infected with RRV, infectious RRV could be isolated from extraintestinal sites, including liver, pancreas, spleen and serum. RRV did not escape the intestine in 4-6-week-old NOD mice. Interestingly, the virus in the pancreas was shown to be present in resident and nonresident macrophages by IF. Infection did not occur in the islets. Infection with RRV did not stimulate the onset of diabetes, but significantly delayed the onset (p = 0.04). Delay of diabetes onset was seen in both 5-day-old and 4-6-week-old orally inoculated mice. These findings indicate that RV infection in children probably does not trigger or exacerbate development of diabetes.[103]
All of the studies described used SCID or normal mice as the model. A single informative paper examined the extraintestinal spread of RV in orally inoculated neonatal rats.[104] An important feature of this study was the perfusion of the animals prior to necropsy, to remove blood-associated virus from the tissue. RRV antigen was detected in the liver in 26 out of 55 (47%) pups at times from 24-96-h postinfection (hpi). Replication in the liver was indicated by isolation of infectious virus from ten out of 12 livers tested. RRV induced liver pathology, with acute inflammation noted in portal tracts and bile ducts, together with microvesicular steatosis, necrosis and inflammatory cells in the liver parenchyma. RRV replication was demonstrated in hepatocytes surrounding the central vein and portal tracts. HAL1166 (human virus) antigen was found in the liver in six out of 15 pups late after infection (other tissues were not examined). Infectious virus was isolated from three out of the six livers; histopathology was not performed. RRV antigen was detected in 14 out of 55 (25%) lungs isolated 48-72 hpi, and infectious virus was isolated in five out of nine lung samples. The lungs of infected animals showed infiltration of inflammatory cells identified morphologically as macrophages, and RRV replication was observed in interstitial macrophages and pneumocytes. RRV antigen was found in 11 out of 30 (37%) spleens early (24-72 hpi) but 17 out of 25 (68%) spleens late after infection (96-216 hpi). In the kidney, RRV antigen was found in 19 out of 55 (36%) pups. Infectious virus was isolated from the kidney samples with the highest antigen levels. RRV antigen could not be detected in the brain of five animals examined. Among the other tissues examined, RRV antigen was sporadically found at low levels in the bladder (25% of animals), thymus (10%), heart (25%), pancreas (25%) and urine (25%). Infectious virus could be isolated from all the samples except urine, but no pathology was noted.[104] This study confirms and extends the studies in mice. Once again, RRV appeared to be particularly hepatopathogenic in the rat, and the lung inflammation recalled the respiratory involvement reported infrequently in children.[105]
In addition, the experiments with rats showed that neither passively acquired transplacental or lactogenic immunity nor active immunity from a prior infection protected pups from extraintestinal spread of either RRV or HAL1166 to the liver of orally inoculated pups as determined by antigen detection.[104] These observations, although preliminary, suggest that vaccination of children may protect against severe diarrhea but not against extraintestinal spread of virus.
One study suggested the spread of RV in a nonhuman primate. Macaque virus strain TUCH was found in low copy number in the CSF of one out of six (17%) macaques that had RV diarrhea from oral infection. The virus was in such a low copy number in the CSF that detection required the use of nested RT-PCR. However, the amplified fragment was shown by sequence to be derived from the infecting TUCH virus strain.[106]
Evidence for Infection of Immune Cells in Animal Models. A number of experimental infection studies have reported the presence of RV in cells of the immune system. In some studies, the presence of virus was not linked to viral replication but in other cases replication in the immune cell was demonstrated ( Table 3 ). In a study of RV interaction with the gut-associated lymphoid system, EDIM virus antigen was found in Ia+/Lyt-1-/Lyt-2-/MAC-1- cells (dendritic) of PP and the MLN early following oral inoculation of mice. Virus replication at these sites was not determined.[86] Subsequently, EDIM antigen, but not RRV antigen, was found sequentially in PP and MLN of 14-day-old mice after oral inoculation.[107] The antigen-positive cells in PP and the MLN were shown by staining for surface markers to be primarily (65-71%) macrophages (CD11B+), although a small proportion of B cells (CD45R+) were antigen positive. No RV antigen was detected in T cells or dendritic cells (DCs). Replication was not demonstrated in any cell type. The authors proposed that virus could travel in macrophages to extraintestinal sites.[107] Further interaction of RV with the lymphoid system was demonstrated by showing that oral infection of mice with strain EC resulted in hyperplasia of PP and the MLN, but not the spleen, early following infection.[108] Increased size of PP and MLN was shown to result from large increases in the numbers of activated B cells, but not T cells, in the tissues. The B-cell activation was T-cell independent because it occurred in T-cell receptor knockout mice. The presence of viral antigen or replication was not examined in this study.[108] Later, it was shown that B-cell activation required RV protein VP7, but that neither viral RNA nor replication was required.[109] In studies detecting virus replication by ssQRT-PCR, virus in the MLN was replicating primarily in DCs (CD11b+), although some replication in B cells and macrophages could not be ruled out.[102] In the rat model, replicating RV was found in macrophages in the lungs and blood vessels.[104] In orally inoculated NOD mice, infectious RRV was found to spread to the pancreas.[103] IF studies showed that in the pancreas, the virus was in macrophages outside the islets and the islet cells were not infected. Furthermore, this study is the first to report the presence of virus in the cellular fraction of the blood. The cellular fraction of blood was treated to lyse red blood cells, and low-titer virus was found in the leukocyte fraction.[103]
Most of these studies are of a preliminary nature but they emphasize the interaction of RV with cells of the gut-associated lymphoid system early after infection. It remains unclear which immune cell type supports replication of RV. Macrophages and DCs are prime suspects, and the migratory nature of these cells suggests that they could carry infectious virus to the blood and to specific peripheral organs.[100,107] The possibility of infection of DCs fits well with the observation that RV strain RRV replicates in 3-46% of mature DC derived from a subset of human volunteers,[110,111] and that RVs activate DCs[112] and induce interferons in DCs.[113]
Studies on RV Antigenemia & Viremia
Antigenemia & Viremia in Animal Models. As noted previously, infectious RV was detected in blood in the earliest studies of EDIM virus (1958), using a sensitive transmission of infection assay.[50] Some 23 years later, antigenemia was documented in calves using ELISA of serum.[83] However, these observations were set aside as work concentrated on the intestinal disease. The modern return to antigenemia and viremia, as topics of study, dates from 2003, when RV infection was shown to extend beyond the intestinal tract and be manifested by antigenemia and/or viremia in children and animal models.[52] This study described viremia in adult (82%) and suckling (100%) mice orally inoculated with murine RV strain EC. Viremia was demonstrated by the infectious nature of serum orally fed to naive animals. In addition, antigenemia was found in 100% of adult and suckling mice orally inoculated with EC or EDIM using ELISA of serum. These results were extended by documenting antigenemia in 100% of orally inoculated suckling rats, adult rabbits and calves. In the latter cases, infectivity of serum was not examined. This study also indicated antigenemia occurred in children (see later and Table 4 ).[52]
Detailed examination of antigenemia and viremia in RV-infected mice confirmed that antigenemia occurred in mice orally infected with murine virus strains EC and EDIM, and with the monkey strain RRV.[114] In addition, RNA was found in 60% of EC-infected mice and viremia was found in 100% of the subset examined for serum infectivity. Viremia was low titer, as tenfold dilution of serum reduced serum transmission of infection to 17%. The viremia was found in the plasma but not the cellular fraction of blood, indicating that the virus was present in infected cells at undetectable levels or not at all. QRT-PCR revealed that RNA was present in serum at a relatively low copy number in positive animals (0-1.5 × 103 copies/µl in serum compared with 2.4 × 105-2.4 × 108 copies/µl in feces). In CD-1 mice, antigenemia was noted for all virus strains examined, following high- or low-dose inoculation (10 or 105 ID50 for each strain). Antigenemia followed virus shedding by approximately 1 day, but persisted as long as shedding in the stool. In addition to CD-1 mice, mouse strains CF-1, BALB/c, C57BL/6 and 129 showed equivalent antigenemia, suggesting that host genetics did not influence development of antigenemia. Age was also not a factor, as both adult and suckling mice developed antigenemia.[114]
The finding of frequent antigenemia and viremia in mice was confirmed in a study using ssQRT-PCR to detect extraintestinal EC or RRV in orally infected BALB/c mice.[102] Negative-sense, but not excess positive ot negative sense, viral RNA was found in the blood, indicating that the virus did not replicate in the blood at detectable levels. Viral RNA was found in the blood of 16 out of 55 (29%) mice orally inoculated with EC and six out of 55 (11%) inoculated with RRV and examined for 15 days postinfection. Similar copy numbers were present in positive animals with both viruses at all times except day 5 when EC inoculated animals had a significantly higher titer. These studies confirmed that viral RNA was plasma associated as the majority was in plasma (~1.1 × 104 copies) as compared with the cellular fraction (~1.5 × 102 copies). An interesting observation in this study was that antigenemia and RNA in the blood did not correlate with virus titer in the gut, as both EC and RRV showed similar levels of antigen and RNA in the blood, but EC replicated approximately 4 log10 more than RRV in the gut. Additionally, the antigen titer and RNA copy number in several of the tissues was higher than in the blood, indicating that replication occurred in the tissue and virus was not simply present in the blood within the tissue.[102]
Preliminary findings[54] of antigenemia and viremia in suckling rats were also confirmed.[104] Antigen was found in the blood of 50 out of 50 (100%) animals orally infected with RRV during the first 4 days of infection, and in ten out of ten(100%) of the animals orally infected with human strain HAL1166 over the first 2 days of infection. Additionally, infectious virus was found in the blood of animals infected with either virus by fluorescent focus formation (FFA). A notable finding was that oral inoculation of RRV caused antigenemia in 50% of animals at a lower dose (1.8x103 pfu/pup) than the dose causing diarrhea in 50% of animals (7.7x105 pfu/pup).[104] This observation suggests that extraintestinal spread of RV may occur in the absence of diarrhea, and that RV-induced systemic symptoms may be difficult to associate with RV infection.
Studies with the human RV strain Wa in gnotobiotic piglets, demonstrated 100% antigenemia and RNA in blood of animals inoculated with virulent Wa but in none of the animals inoculated with attenuated Wa.[115] Antigenemia occurred regardless of whether the virus was inoculated intranasally, by mouth, or by gavage, although viral shedding in the feces was approximately 1 log10 lower with the attenuated virus. That viremia occurred in the piglets was demonstrated in naive piglets fed with pooled serum from antigenemic animals. All recipient piglets developed diarrhea and antigenemia, whereas none of the piglets fed pooled serum from mock or attenuated virus infected pigs became diseased or developed antigenemia.[115]
In a small study, RV RNA was detected by PCR in the serum of two out of six (33%) juvenile macaques orally inoculated with macaque RV strain TUCH. Real-time PCR revealed very low levels of RNA in the plasma (≤20 copies/µl) that were near the limit of detection. Application of nested PCR and sequencing confirmed the presence of TUCH RNA in the plasma.[106]
Antigenemia & Viremia in Children. Efforts also turned to the demonstration of viremia in RV-infected children. The demonstration of viremia is quite difficult since the infectiousness of serum cannot be detected by inoculating naive children, and because human RVs are difficult to detect by plaque assay or FFA immediately after isolation from children. To date, antigenemia and the presence of viral RNA in blood has been documented several times, although the occurrence of viremia was only demonstrated recently.
The first indication that RV antigens could enter the blood of children came from a retrospective study in which 66% of children positive for stool RV had antigen in the serum and 50% had serum RNA.[54] Detection of RNA in the serum by RT-PCR allowed sequence analysis and demonstration that at least two different G serotypes (G1 and G4) were capable of establishing viremia. Infectivity of serum from children could not be tested.[54] The high frequency of antigenemia following the natural (oral) route of infection stimulated considerable interest in the question of extraintestinal spread of virus during RV infection.
A study performed during an outbreak of acute gastroenteritis in Jamaica revealed the presence of RV antigen in the serum of 30 out of 70 (43%) RV stool-positive children, and only one out of 53 (2%) controls.[116] PCR demonstrated that high levels of stool antigen correlated with RNA in the serum. In addition, serum antigen levels showed a negative association with time between symptom onset and the time of the acute serum sample. High serum antigen levels correlated significantly with primary infection when acute serum RV IgG titers were less than 25 compared with secondary infections (acute serum RV IgG > 25) where antigenemia levels were lower. Antigenemia did not associate with virus strain as P[8]G1 (61%), P[8]G9 (29%) and mixed infections (7%) all resulted in detectable antigenemia. A tendency of severity of illness to associate with high levels of antigenemia was noted.[116]
Another study examined the antigenemia in children with encephalopathy accompanied by RV gastroenteritis in Japan.[117] Eight children who had an encephalopathy with concurrent RV in their stool were identified. Five out of eight (63%) had RV antigen in acute-phase sera but none had antigen in convalescent sera. In this study, RV RNA could not be detected in the serum by PCR. Interestingly, the three children with no antigen or RV RNA in serum had RV RNA detectable by PCR in the CSF.[117]
A small prospective study in Italy detected RV RNA in the serum of nine out of 14 (64%) of children with RV-positive stools.[118] The presence of RNA in the serum showed a high degree of association with fever greater than 38.5°C and abnormal serum ALT levels. However, no severe extraintestinal disease was noted.[118]
A retrospective study of serum samples from India found antigenemia in 65 out of 102 (64%) of RV-infected children.[119] Only 3% of control children with non-RV diarrhea had antigenemia. High levels of fecal RV antigen correlated significantly (p < 0.01) with antigenemia. RV RNA was detected in 26 out of 28 (93%) antigenemic children. RNA copy numbers ranged from 4.0 × 104 to 4.0 × 106 copies/ml of serum. However, fecal and serum RNA copy numbers showed no correlation. Blood antigen levels also had a negative correlation (p < 0.03) with the acute RV IgG titers, indicating that pre-existing antibodies lowered the level of blood antigen. In the 65 samples for which the G serotype was determined, those with G1 infections had antigenemia more often (85%) than those with other serotypes or mixed infections. Antigenemia did not correlate with the severity of disease.[119]
Viremia in children was detected in a very recent study.[57] In this prospective study, 57 out of 98 (58%) children with gastroenteritis had RV in the stool. Among the RV-positive stool group, 51 out of 57 (90%) had serum antigenemia. A total of zero out of 45 controls were positive for stool or serum antigen. Further analysis revealed five out of 41 (12%) children with gastroenteritis but RV antigen-negative stool had serum antigenemia, suggesting that antigenemia may occur in the absence of antigen shedding. Two of the antigenemic children had at least a fourfold rise in serum antibody titer, indicating they were indeed RV infected. To examine the possibility that antigenemia could occur in children not thought to be RV-infected, children with bronchiolitis not caused by a panel of respiratory pathogens and without diarrhea were examined. Two out of seven in this group had serum antigenemia. A subset of children was examined for viremia; 11 out of 11 (100%) with serum antigen had detectable infectious RV in the serum. Two out of nine (22%) children who were serum antigen negative but stool antigen positive had infectious virus in the serum. Limiting dilution experiments revealed infectious virus could be detected in serum at 1:80 or 1:160, indicating that the titer in the blood is low.[57] Additional analyses showed no correlation between serum antigenemia and time of collection of blood (within the first week), age of the child or viral serotype. A significant negative correlation was found between RV-specific serum IgA and IgG titers and serum antigenemia. Antigenemia was predictive of viremia in children (p = 0.002).[57] This study was the first to conclusively demonstrate viremia in children.
A Model for Systemic Spread of RV
The observations described previously allow speculation as to the mechanism of RV spread to circulation and peripheral tissues. Key among these observations is that the highest titers of RV outside the GI tract are found in the MLN.[102] This observation, coupled with the lymphatic route of spread described in mice,[100] suggests that RV initially escapes the GI tract via the gut-associated lymphoid system (Figure 2). Indeed, a lymphatic mode of spread has been proposed.[100,107]

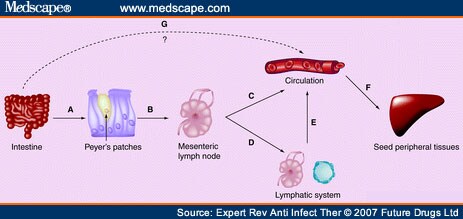
Figure 2.
Model for systemic spread of rotavirus. (A) Virus crosses M cells and infects a migratory cell (macrophage or dendritic cell) in the underlying Peyer's patch. (B) Virus replicates in the migratory cell, which migrates to the mesenteric lymph node (MLN). (C) In the MLN, the infected migratory cell may release virus into circulation, resulting in viremia. (D) In the MLN, the infected migratory cell may continue to migrate through lymphatics. (E) Cell-associated virus in the lymphatics enters the portal system which dumps into the circulation. (F) In the periphery, infected migratory cells may exit circulation and home to specific organs where the virus is released and infects cells locally within the tissue. Alternatively, free virus in circulation may adsorb to tissue-specific receptors on endothelial cells, infect those cells and gain entry to the tissue from the basal side of the endothelial cells. (G) As a result of damage in the intestine, the virus may directly enter circulation.
The initial step in escape from the gut is most likely the passage of virus particles through the microfold (M) cells of the dome epithelium that overlays PP lymphoid folicles (Figure 2A). Reovirus is a prime example where virus crosses M cells[120] for presentation to the immune system. Unlike reovirus, which is processed for antigen presentation, it appears that RV actively infects a migratory cell next to PP. Support for this notion comes from the observation that RV replication was reported in DCs, and potentially B cells and macrophages in mice,[102] in lung macrophages in rats[103] and cultured myeloid DCs from humans.[110] Furthermore, in genetic experiments, escape from the gut segregated with a NSP,[53] which implies infection is necessary for escape. The infected migratory cell is then free to migrate through the lymphatics to the MLN.
Once the infected migratory cell has reached the MLN (Figure 2B), the infection may spread to other cells that are encountered at that site. Support for the notion of amplification of infection in the MLN comes from the finding of the highest replicating viral load[102] and the highest titers of infectious virus[100] in the MLN of any tissue outside the GI tract. It appears that the MLN may represent a decision site relative to subsequent spread, as genetic experiments indicate that among viruses that reach the MLN, some can spread no further.[100] The infected migratory cell may release virus in the MLN that enters the circulation to establish viremia (Figure 2C), the migratory cell may enter circulation (Figure 2C) or the migratory cell may continue to traverse the lymphatic system (Figure 2D) and ultimately enter circulation through the portal system (Figure 2E).
It is unclear how virus leaves circulation or the lymphatic system to establish infection in peripheral tissues. The majority of virus in the circulation is free in the plasma.[102,114] This free virus may be recognized by tissue-specific receptors on endothelial cells and be taken into the peripheral tissue that way. Virus associated with infected migratory cells may be taken into the tissue by homing mechanisms inherent to the infected cell. Alternatively, virus associated with infected migratory cells in the lymphatic system may enter circulation through the portal system and follow the pathways for cell-associated virus in the blood, or the infected migratory cell may leave the lymphatic system to establish infection directly in the tissue to which it moves. It is not currently understood whether infection is initiated by cell-free virus that enters the peripheral site or the virus first enters the peripheral site in an infected migratory cell.
An alternative mechanism of RV entry into the circulation is directly from the basal aspect of the infected enteric epithelium. RV is not released from the basolateral aspect of polarized epithelial cells. However, RV infection of polarized epithelia abolishes transepithelial resistance, increasing paracellular leakage[121] and provides a pathway by which virus could reach the basolateral surface of adjacent cells for infection. Virus at the basolateral location could also cross the basement membrane for entry directly into the circulation (Figure 2G). It is clear that the enterotoxin, NSP4, has the potential to enter the circulation by this mechanism.[122]
Expert Rev Anti Infect Ther. 2007;5(4):591-612. © 2007 Future Drugs Ltd.
Cite this: Systemic Rotavirus Infection - Medscape - Aug 01, 2007.








Comments