Materials and Methods
This study was conducted as a phase IIIb, large-scale, randomised, multicentre, double-blind, prospective, active-controlled clinical trial. The institutional review board of each participating study site approved the protocol prior to study initiation. Each patient provided written informed consent prior to initiation of any study-related procedures.
Patients who were at least 18 years of age with a history of at least two episodes of heartburn per week that partially responded to treatment with a PPI, H2 receptor antagonist or antacids within the 30 days prior to screening were eligible for enrollment in the pretreatment period. Pregnant or lactating women were excluded from study participation, as were patients with a known allergy to any PPI.
Patients meeting the heartburn criteria were required to discontinue their use of prescription or nonprescription (over-the-counter) medication(s) to treat heartburn (e.g. PPIs, H2-receptor antagonists, antacids, sucralfate, prokinetics and anti-cholinergic agents) in order to participate in the 7-day pretreatment screening period. Patients were dispensed antacid tablets (Gelusil ® 1 ; Pfizer Consumer Health Care, Morris Plains, NJ, USA) and allowed to use them during the pretreatment period for 'breakthrough' heartburn. Patients were to complete, on a daily basis for 7 days, a prompted interactive voice response system (IVRS), or a paper diary if IVRS was inaccessible, to record the presence of and the worse severity for any daytime and/or night-time heartburn experienced on the previous day. The severity of heartburn was recorded as 'none', 'mild' (occasional heartburn, can be ignored, does not influence daily routine or sleep), 'moderate' (heartburn cannot be ignored and/or occasionally influences daily routine or sleep), 'severe' (heartburn present most of the day or night and/or regularly influences daily routine or sleep), or 'very severe' (constant heartburn and/or markedly influences daily routine or sleep).
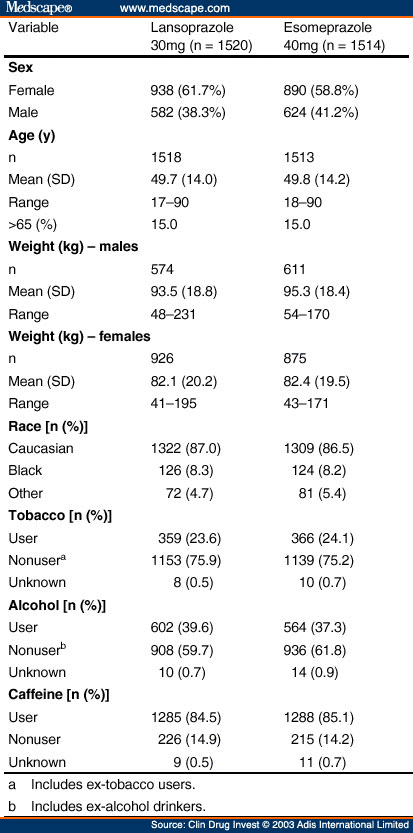
Patients who experienced day and/or night heartburn of at least mild severity occurring at least 50% of the days and at least one episode of moderate, severe or very severe daytime and/or night-time heartburn during the pretreatment period were eligible for randomisation in the double-blind treatment period. Prior to study medication randomisation, patients underwent a complete medical and social history that included a gastrointestinal symptom history and assessment of smoking, caffeine and alcohol consumption, as well as documentation of all medications (prescription or nonprescription) taken within the prior 30 days. A complete physical examination was also performed, including recording of blood pressure, respiratory rate, body temperature and body-weight. Women of childbearing potential had to have a negative pregnancy test prior to treatment randomisation.
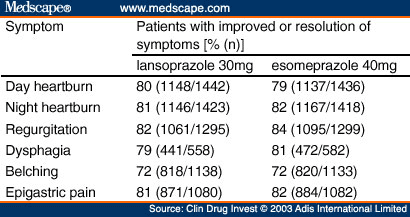
A GERD symptom assessment was performed by the investigator at the end of the pretreatment period with symptoms of day heartburn, night heartburn, acid regurgitation, dysphagia, belching and epigastric pain, as defined in Table I , that occurred during the prior 7 days. The severity of each symptom was classified as 'none' (no symptoms), 'mild' (symptom does not last long and is easily tolerated), 'moderate' (symptom causes discomfort and interrupts usual activities including sleep), or 'severe' (symptom causes great interference with usual activities and may be incapacitating [including sleep]).
Patients with symptomatic GERD based on pre-set criteria were assigned a unique patient number which encoded the randomisation in a 1 : 1 ratio to one of the two treatment groups (lansoprazole 30mg once daily [Prevacid ® ; TAP Pharmaceutical Products Inc., Lake Forest, IL, USA] or esomeprazole 40mg once daily [Nexium ® ; AstraZeneca Pharmaceuticals LP, Wilmington, DE, USA]) for 2 weeks. Both study medications were made to appear identical by overencapsulating in iron-gray opaque capsules to maintain the study blind for the patients, investigators, study personnel and sponsor. In vitro dissolution studies confirmed that blinded capsules had a similar profile to unblinded capsules. Patients were instructed to take the study medication each morning before breakfast. They were instructed to use the IVRS to report the presence and maximum severity (described as none, mild, moderate, severe or very severe according to the above-mentioned GERD assessment definitions) of the previous day's daytime and night-time heartburn as well as the self-administration of study medication. Patients returned for the follow-up visit at the end of the 2-week treatment period.
All patients were contacted by telephone on study day 1 to confirm that they had taken the first dose of study medication as instructed. Additional telephone contacts were performed at study day 7 and as needed to confirm that the patient continued to be compliant with study drug administration and the daily IVRS completion and to assess any adverse events.
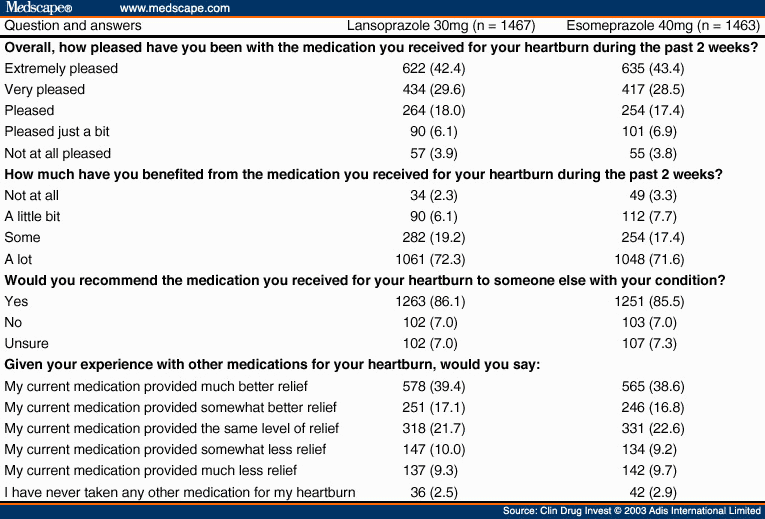
At the week 2 follow-up visit, patients underwent a repeat assessment of vital signs and GERD symptoms by the investigator. Drug accountability was performed to assess compliance, and a review of concurrent medication use was performed. Patients were asked about their treatment satisfaction using the following questions.
Overall, how pleased have you been with the medication you received for your heartburn during the last 2 weeks? (Possible responses: extremely pleased, very pleased, pleased, pleased just a bit, not at all pleased.)
How much have you benefited from the medication you received for your heartburn during the past 2 weeks? (Possible responses: not at all, a little bit, some, a lot.)
Would you recommend the medication you received for your heartburn to someone else with your condition? (Possible responses: yes, no, unsure.)
Given your experience with other medications for your heartburn, would you say your current medication provided... ? (Possible responses: much better relief, somewhat better relief, the same level of relief, somewhat less relief, much less relief, I have never taken any other medication for my heartburn.)
The tolerability and safety of the treatments was determined by systematic assessment of patient-reported and investigator-observed adverse events. Adverse events were rated in severity as mild, moderate, or severe and classified as to their relationship to study drug as definite, probable, possible, unlikely or not related. The investigator followed all patients experiencing an adverse event until there was a return to baseline or clinically satisfactory resolution was achieved.
A sample size of 3000 patients (1500 in each treatment group) was planned. This sample size was selected to have at least a 95% power to detect differences between lansoprazole and esomeprazole at the 0.0025 (two-tailed) level, assuming that the mean (and standard deviation) for the percentage of days with heartburn over the first 3 days of treatment would be 48% (43%) for patients treated with lansoprazole and 56% (41%) for patients treated with esomeprazole, using normal approximations. A p-value of 0.0025 was chosen to demonstrate that the strength of the efficacy results would be equivalent to that of efficacy results obtained in two independent studies using a p-value of 0.05.
Data were analysed using the intent-to-treat approach. This analysis included all patients who entered the study with heartburn recorded during the pretreatment period, received at least one dose of study drug, and completed any daily IVRS response regardless of any protocol deviation. For baseline characteristics, the Chi-square test was used for the categorical variables, and a one-way analysis of variance (ANOVA) was used for the continuous variables.
The primary efficacy analyses were the comparison of heartburn symptom relief rates over the first 3 days of treatment with lansoprazole 30mg once daily or esomeprazole 40mg once daily as measured by the percentage of days with heartburn and the percentage of nights with heartburn, using data from the IVRS completed on diary days 2, 3 and 4 that recorded symptoms experienced on days 1, 2 and 3. Diary day 1 is defined as the first day that the patient took study drug as recorded in the IVRS. Treatment comparison of the primary efficacy endpoint was performed using the Cochran-Mantel- Haenszel test with investigator as the stratum.
Additional efficacy endpoints included the proportion of patients without daytime heartburn or without night-time heartburn, and the average severity of daytime heartburn and night-time heartburn experienced on the first treatment day and during the first 3 days, first week and 2-week treatment period. During the first week and the 2- week treatment period, the percentage of days with heartburn and the percentage of nights with heart-burn were also determined. The proportions of patients without heartburn over each of the evaluated treatment periods were compared between the treatment groups using the Cochran-Mantel-Haenszel test, with the investigative site as the stratum. An average heartburn severity score across days during treatment was calculated on the basis of: 0 = none, 1 = mild, 2 = moderate, 3 = severe and 4 = very severe. Treatment comparison of the additional efficacy endpoints of the percentage of days and nights with heartburn and the average severity of day and night heartburn was performed using the van Elteren method of combining the Wilcoxon test statistics,[18] with investigator site as the stratum. Ad hoc analyses of comparisons between the treatment groups for the percentage of days with heartburn and the percentage of nights with heartburn, as well as average severity of day-time and night-time heartburn, were performed stratifying for pretreatment daytime and night-time heartburn severity, respectively, using the van Elteren method. The average pretreatment daytime or night-time heartburn severity score was categorised using 0-1.0 = mild, >1.0-2.0 = moderate, >2.0-3.0 = severe and >3.0-4.0 = very severe, and used as the stratum. The cumulative percentage of patients who achieved the start of sustained resolution of heartburn (defined as 7 consecutive days with no heartburn) was calculated for each treatment group, and the time to sustained resolution was compared between treatment groups using the log-rank test.
The week 2 GERD symptom investigator assessment and the patient's treatment satisfaction assessment were analysed with Cochran-Mantel-Haenszel methodology for ordered responses using the baseline values as the stratum.
Comparisons between the treatment groups for treatment-emergent adverse events were performed using Fisher's exact test. Changes in vital signs from baseline to the final treatment visit were compared between the regimens by ANOVA.
Clin Drug Invest. 2003;23(2) © 2003 Adis Data Information BV
Use of tradenames is for product identification only and does not imply endorsement.
Cite this: Lansoprazole and Esomeprazole in Symptomatic GERD - Medscape - Feb 01, 2003.