Abstract and Introduction
Introduction
In subjects with type 1 diabetes, autoimmune destruction of pancreatic β-cells leads eventually to an absolute requirement for insulin replacement therapy. Insulin delivered exogenously is not subject to normal physiological feedback regulation, so it may induce hypoglycemia even in the presence of an intact counterregulatory response. The average individual with type 1 diabetes experiences about two episodes of symptomatic hypoglycemia per week, a figure that has not changed substantially in the last 20 years.[1] Severe hypoglycemia (requiring help for recovery) has an annual prevalence of 30–40% and an annual incidence of 1.0 – 1.7 episodes per patient per year.[1] This risk is increased markedly with the increasing duration of the disease and strict glycemic control. In subjects with type 2 diabetes, the increasing duration of the disease and the more widespread use of insulin therapy also increase the risk of severe hypoglycemia. This was reflected in a recent survey in Tayside, Scotland, which found the proportion of severe hypoglycemic episodes needing emergency medical assistance was similar between type 1 and insulin-treated type 2 diabetic patients.[2]
The experience of hypoglycemia is not limited to a transient impairment of cognition. We now recognize that hypoglycemia carries with it a recognized morbidity and mortality[3] and creates a negative mood-state characterized by reduced energy and increased tension.[4] This may explain why hypoglycemia is greatly feared by individuals with diabetes; so much so that the fear of hypoglycemia is rated with the same degree of concern as the development of sight-threatening retinopathy or end-stage renal disease. This fear of hypoglycemia influences an individual's ability to adhere to optimal insulin replacement regimens and to put in place those measures required to achieve near-normal glucose control. In this way, hypoglycemia has emerged as a major obstacle to achieving the goals of intensive insulin therapy in everyday clinical practice.
In this review, we briefly describe the primary defects in hypoglycemia counterregulation, which are almost universally present in individuals with type 1 diabetes and, within this context, subsequently provide a general overview of the current state of research into the more basic mechanisms underlying the detection of hypoglycemia. Our focus tends to be on research into animal models, reflecting the focus of recent activity in this area. Animal models are a valuable tool for dissecting the molecular mechanisms involved in glucose sensing. To date, most animal models seem to show a similar hierarchy of responses to acute hypoglycemia—as well as developing similar defects to repeated hypoglycemia—as their human counterparts.[5] However, species differences, particularly in brain metabolism given the unique size and metabolic demands of the human brain or in islet substructure, mean that data gleaned from animal studies still require further validation in human subjects before they can be confidently translated into clinical practice. A recent clinical review of hypoglycemia research[6] provides a detailed discussion on the clinical trials in human subjects.
Abnormal Glucose Counterregulation in Diabetes
In nondiabetic individuals, hypoglycemia initiates a classic negative feedback counterregulatory response in which the fall in glucose leads to a series of neurohumoral and behavioral responses designed to restore normal glucose levels. Key steps in this homeostatic response are the suppression of endogenous insulin secretion and a stimulus to the secretion of the counterregulatory hormones, glucagon and epinephrine, which act rapidly to stimulate endogenous glucose production and to limit peripheral glucose utilization, thus increasing glucose delivery to the brain.
Three major defects in this homeostatic response contribute to the high frequency of hypoglycemia in type 1 diabetes. Firstly, the loss of β-cell insulin secretion and the need for exogenous insulin therapy mean that hypoglycemia is more likely to develop because of unregulated and sustained hyperinsulinemia. Secondly, within 5 years of disease diagnosis, almost all individuals with type 1 diabetes fail to generate an adequate glucagon response to hypoglycemia.[7] Glucagon is the principal rapid-acting counterregulatory hormone, and the portal insulin-to-glucagon ratio is the major determinant of hepatic glucose production. Reduced or absent glucagon release results in a marked impairment of glucose recovery from hypoglycemia.[8] A number of intra- and extra-pancreatic factors are thought to contribute to this defect. Briefly, a failure in local regulation of β- to α-cell signaling by insulin, zinc, and possibly the neurotransmitter γ-aminobutyric acid (GABA) during hypoglycemia probably play the dominant role in the genesis of this defect, particularly as it seems to track with the progressive loss of β-cell function.[9] However, recent data suggest that the inhibitory effect of exogenous insulin on α-cell glucagon release is in part mediated at the level of the ventromedial hypothalamus (VMH).[10] Thus, the loss of glucagon response to insulin-induced hypoglycemia in C-peptide–deficient type 1 diabetic patients may to be due to the simultaneous increase in insulin levels both within the islet and the VMH. In addition, evidence exists of a local intra-islet sympathetic neuropathy,[11] which may contribute in part to impaired glucagon release during hypoglycemia. Again, species differences in islet substructure in rodents limit our translation of these findings to human physiology, but the recognition that a number of defects may contribute to the loss of α-cell glucagon secretion during hypoglycemia opens up the possibility of novel therapeutic approaches, such as stimulation of central nervous system (CNS) sensing mechanisms. The selective inability of the α-cell to respond appropriately to a hypoglycemic challenge is a hallmark of type 1[8] and long-duration type 2 diabetes,[12] which remains poorly understood. Thirdly, the major defect in the counterregulatory response to hypoglycemia in diabetes is a reduced autonomic response. This affects the majority of individuals with type 1 diabetes by 10 years disease duration.[7] Hypoglycemia normally leads to activation of the autonomic nervous system resulting in increased hepatic glucose production and reduced glucose uptake in peripheral tissues. In liver stimulation, sympathetic nervous system activation increases both glycogenolysis and gluconeogenesis; the latter via a simultaneous increase in the delivery of gluconeogenic substrates and free fatty acids.[13] The autonomic response is closely associated with the generation of a symptomatic response to hypoglycemia and, as such, when this response becomes impaired there is usually reduced awareness of hypoglycemia as well as a reduction in catecholamine release. This association means the autonomic response to hypoglycemia is critically important in individuals with type 1 diabetes. As will be discussed later in this review, a defective autonomic counterregulatory response results primarily from prior exposure to hypoglycemia per se,[14] a situation that occurs most frequently during intensive insulin therapy. This sets up a vicious cycle whereby hypoglycemia increases the likelihood of subsequent hypoglycemia.
Thus, the glucose counterregulatory defense against hypoglycemia in individuals with diabetes becomes impaired at almost every level and rendered even more defective through intensive insulin therapy. In the following sections, we will examine some of the basic mechanisms underlying the detection of hypoglycemia. In vivo and ex vivo animal models have been used to ask the questions, where does the body sense fluctuations in glucose levels, how is glucose sensed, and why does this mechanism become impaired following recurrent hypoglycemia?
The Molecular Biology of Hypoglycemia Detection
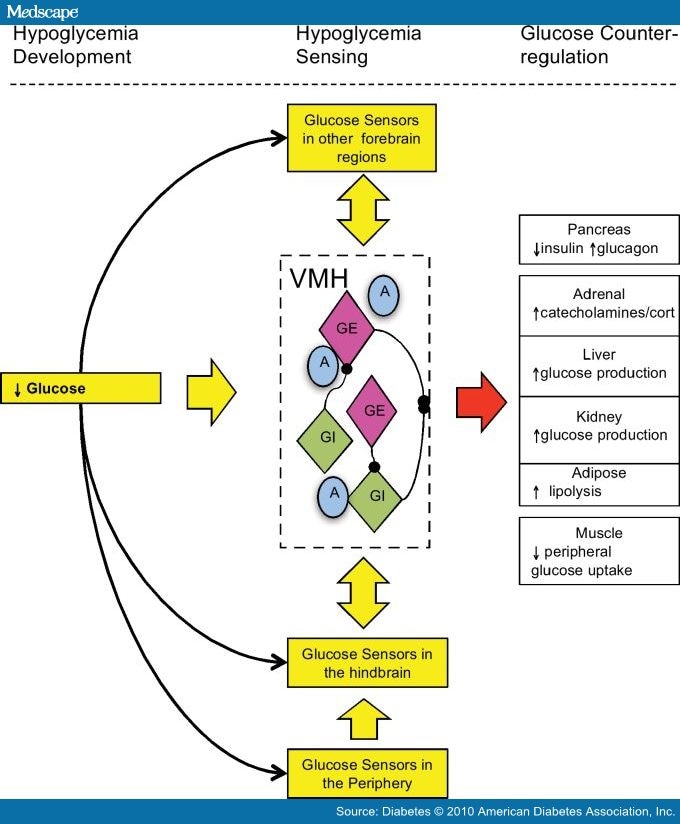
Hypoglycemia Sensors. It is currently believed that hypoglycemia is detected by specialized cells/neurons located within discrete regions of the CNS and periphery, and it seems likely that these cells are linked together in some way providing an integrated mechanism for monitoring whole-body glucose and/or fuel homeostasis (Fig. 1). In an excellent recent review of this integrative glucose-sensing network, Watts and Donovan[15] describe how peripheral, hindbrain, and hypothalamic glucose sensors form a classical sensory-motor integrative pathway. They illustrate how forebrain integrative networks might modify hindbrain glucose-sensing autonomic reflex loops. This model could explain how different stressors (e.g., hypoglycemia and exercise) might interact, and why defective hypoglycemia counterregulation could arise through defects in these forebrain integrative networks.
Figure 1.
Integrative model of hypoglycemia detection. A falling glucose is detected by peripheral and central glucose-sensing cells/neurons. Peripheral glucose sensors signal back to glucose-sensing regions of the hindbrain in turn activating efferent pathways that initiate a counterregulatory response. At the same time, glucose sensors in the hypothalamus, such as those in the VMH and other forebrain regions, detect falling glucose, also activating efferent pathways that initiate a counterregulatory response. Integrative pathways between hindbrain, hypothalamic, and other forebrain regions are reciprocally connected and can modulate responses to the hypoglycemic signal. Glucose-sensing regions in the brain, such as the VMH, contain GE and GI neurons and an astrocytic support structure. A, astrocytic.
To date, glucose sensors in the periphery, apart from the pancreatic β-cell, have been found in the intestine, hepatoportal vein, and carotid body.[7] Within the CNS, ex-vivo electrophysiological studies have identified a number of areas in the brain that contain neurons sensitive to local changes in glucose.[7] One brain region in particular, the VMH, appears to plays a crucial role during hypoglycemia and was the subject of a recent review.[16] The specialized glucose-sensing neurons in the CNS have been broadly defined as either glucose-excited (GE), which increases their action potential frequency when glucose rises, or glucose-inhibited (GI), which increases their action potential frequency when glucose levels fall.[17] These neurons are liable to react in a coordinated manner to alterations in the glucose level to which they are exposed. The neurons also respond to other metabolites such as lactate and β-hydroxybutyrate, as well as hormones such as insulin, leptin, and possibly glucagon-like peptide 1, reflecting the central role they play in responding to alterations in fuel supply and in maintaining glucose homeostasis. From an evolutionary perspective, it seems very likely that these neurons have developed to ensure an adequate supply of fuel to the brain during periods of prolonged starvation because of the limited capacity of the brain to store fuel in depots such as glycogen or fat. In this context, the ability to integrate many different aspects of human metabolism is essential to ensure a continuous supply of glucose to the brain.
Hypoglycemia Sensing
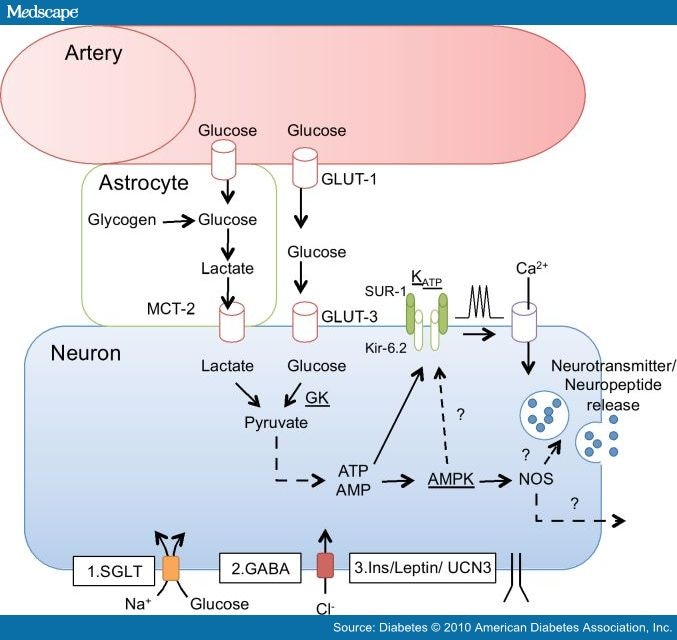
The Hypothalamic β-cell. A number of mechanisms may underlie glucose sensing by GE/GI neurons in the CNS. These are not necessarily distinct or redundant mechanisms and, at least in the authors' opinion, probably all play some role in the detection of hypoglycemia. Most studies indicate that the principal glucose-sensing mechanism within these specialized neurons parallels that used by the pancreatic β-cell, namely in the critical roles for glucokinase (GK),[18] the ATP-sensitive potassium channel (KATP),[19] and AMP-activated protein kinase (AMPK)[20] (Fig. 2). The pancreatic isoform of GK (the critical regulator of glycolytic production of ATP and KATP channel activity in the pancreatic β-cell) is expressed in the majority of glucose-sensing neurons as is mRNA for sulfonurea receptor (SUR)-1 and Kir6.2 subunits of the KATP channel.[21] Pharmacological or adenoviral manipulation of GK modulates hypothalamic glucose-sensing neurons ex-vivo, and selective down-regulation of GK using RNA interference in the VMH of rats suppresses the counterregulatory response to acute hypoglycemia.[22] In addition, down-regulating GK in primary VMH neuronal cultures using RNA interference leads to the loss of all demonstrable glucose-sensing (GE and GI) activity.[18] Similarly, electrophysiological studies of rat[23] and mouse hypothalamic slice preparations[24] demonstrate that sulfonylureas modulate the firing rate of glucose-sensing neurons, and local in vivo application of a KATP channel blocker to the VMH suppresses, while the opening of the KATP channel amplifies the glucose counterregulatory response to acute hypoglycemia.[7] GLUT-2, the high-capacity, low-affinity GLUT of the pancreatic β-cell, may also play a role in central glucose sensing,[25] although limitations in the transgenic model employed in this study and the difficulty in detecting GLUT-2 in the brain mean this data needs replicating.

Figure 2.
A simplified model of glucose-sensing mechanisms present in the brain. Glucose from the arterial supply is transported either directly into neurons via GLUT-3 (possibly GLUT-2) or indirectly as lactate generated through glycolysis in astrocytes. Glucose is phosphorylated by GK, a key regulatory step in glucose sensing, before undergoing oxidative-phosphorylation to generate ATP. ATP closes the SUR-1–selective KATP, leading to membrane depolarization, calcium influx, and release of neurotransmitters and/or neuropeptides. In addition, AMP-to-ATP ratios are monitored by AMPK, activation that during hypoglycemia stimulates the production of nitric oxide and may act via the KATP channel or directly to stimulate neurotransmitter release. Additional mechanisms that may modulate this basic glucose-sensing mechanism include: 1) SGLTs, 2) GABAergic inhibition (or modulation by other neurotransmitters such as norepinephrine), and 3) the actions of hormones such as insulin and leptin and neuropeptides such as urocortin 3. CI−, chloride; Ins, insulin; MCT, monocarboxylate transporters; NOS, nitric oxide synthase; UCN3, urocortin 3.
Evidence is also emerging for an important role for AMPK in glucose sensing, particularly during hypoglycemia. AMPK is an ancient, highly conserved serine/threonine kinase that is activated during cellular energy depletion and acts to suppress ATP-consuming pathways and to activate ATP-generating pathways. Hypothalamic AMPK is activated in response to fasting or central glucoprivation. During hypoglycemia, local in vivo pharmacological activation of AMPK in the VMH amplifies counterregulatory responses while selective AMPK down-regulation in the VMH suppresses the responses.[7] It has been suggested that AMPK acts as the dominant glucose sensor in GI neurons,[26] however loss of glucose-sensing ability in transgenic mouse models with selective loss of AMPK in classical hypothalamic GE neurons[27] and pancreatic β-cells[28] would seem to suggest that AMPK acts as a functional glucose sensor within both GI and GE neurons.[7]
The "Glucose" Signal. In addition to the classical pathway of glucose sensing, Burdakov et al.[29] have proposed the novel and intriguing hypothesis that glucose, independent of its oxidation, may modulate the action potential in glucose-sensing neurons. Glucose transport into the neuron is thought to be coupled directly with the transmembrane movement of ions, such as those used by sodium glucose cotransporters (SGLTs). Given intracerebroventricular, phloridizin, a nonselective inhibitor of SGLTs, increased food intake in rats and inhibited VMH GE neurons,[29] while α-methylglucopyranoside, a nonmetabolizable substrate of SGLTs, excited GE neurons in primary rat hypothalamic cultures.[30] Alternatively, glucose might bind to an extra-cellular receptor that could alter electrical activity without transporting the glucose into the neuron.[29] Whether glucose sensing in the brain occurs primarily through this mechanism, or whether glucose per se might act to potentiate the signal induced by the oxidation of glucose in sensing neurons, remains to be determined.
Astrocytic Glucose Sensing. Finally, it is well established that within the CNS, astrocytes and neurons (and blood vessels) work together as functional units. Importantly, the cerebral blood vessels delivering glucose to the brain are almost completely surrounded by a network of astrocytic foot processes. This raises the possibility that glucose may regulate sensing neurons at least in part indirectly via astrocytes. Tanycytes, for instance, are specialized astrocytic cells that line much of the floor of the third ventricle and express GLUT-2, GK, and the KATP channel and send long processes that terminate in the VMH. Tanycytes show reversible inhibition by third ventricular delivery of alloxan (taken up through GLUT-2) in a temporal pattern that parallels changes in the hormonal counterregulatory response to systemic 2-deoxyglucose.[31] On the basis of these findings, Sanders et al.[31] have suggested that tanycytes may play a critical role in glucose sensing during hypoglycemia, transmitting the glucoprivic signal to neurons in the VMH, which then stimulate a counterregulatory response.
The current literature, taken together, would appear to suggest that the characteristic feature of glucose-sensing cells is the presence of GK and AMPK. Intriguingly, this would suggest that the glucose-sensing mechanism may be a universal mechanism even if the cell is activated or inhibited by glucose. Downstream signaling and the specific neurotransmitters/neuropeptides released would then determine the output of that glucose signal. It is important to note that this would also imply that a glucose-sensing neuronal population did not need to be directly involved in glucose homeostasis. Neuropeptide Y neurons are GI neurons that play a major role in stimulating food intake in response to glucoprivation, but there is no evidence to date indicating a direct role in the stimulus to hormonal counterregulation.
Hypoglycemia Activation
Once a change in glucose is sensed, the neuron needs to communicate that signal to a downstream neuron in the pathway that eventually leads to glucose counterregulation. In general, neural communication relies on the release of classical neurotransmitters, such as GABA, glutamate, neuropeptides, or unconventional transmitters such as nitric oxide. GAD, the rate limiting enzyme in GABA synthesis, is expressed in 56% of GE and 36% of GI neurons.[21] GABA levels in VMH interstitial fluid are decreased during acute hypoglycemia, and in vivo antagonism of the VMH GABA amplifies the counterregulatory hormone response to acute hypoglycemia.[32] It is important here to note that the source of GABA input to the VMH during hypoglycemia is not as yet known and may arise from surrounding hypothalamic or other forebrain regions. Interestingly, recurrent hypoglycemia leads to a significant increase in GAD65 mRNA and protein (33 and 580%, respectively) in the VMH, while VMH GABA concentrations measured by microdialysis were more than threefold higher, suggesting that recurrent hypoglycemia results in increased VMH GABA inhibitory tone. Increased GABA tone could contribute to reduced action potential frequency in VMH glucose-sensing neurons with the net result of this being to suppress the counterregulatory response during subsequent hypoglycemia.
A single report[33] suggests that the excitatory output from glucose sensors such as the VMH may be glutamatergic, while excitatory input to the VMH from brain stem noradrenergic neurons may link peripheral to hypothalamic glucose sensors.[34] In addition, caudal hindbrain serotonergic neurons express GK and project to sympathetic interomediolateral neurons in the spinal cord.[35] Recently, it was reported that 6- or 20-days delivery of a selective serotonin reuptake inhibitor to normal Sprague-Dawley rats amplified the counterregulatory response to acute hypoglycemia and prevented the development of defective counterregulation in rats exposed to repeated hypoglycemic stress.[35] Interestingly, human subjects with type 1 diabetes also show enhanced counterregulation to hypoglycemia following 6 weeks of selective serotonin reuptake inhibitor therapy.[36] Finally, the unconventional transmitter nitric oxide may also provide a signal to downstream neurons.[37]
Hypothalamic glucose-sensing neurons can also be regulated by local or peripheral release of neuropeptides. Davis et al.[38] was the first to demonstrate this in the context of the hypoglycemic stress response by showing the important regulatory role of systemic glucocorticoids, while Flannagan et al.[39] noted a potential role for systemic corticotrophin-releasing hormone (CRH). More recently it has been shown that VMH urocortin 3, also a member of the CRH family of neuropeptides, has a marked suppressive action on counterregulatory responses to acute hypoglycemia.[40] Conversely, VMH microinjection of CRH, which acts primarily through CRH-receptor type 1, amplifies the counterregulatory response.[41] Thus, there appears to be feedback inhibition to the hypothalamus of the hypoglycemic stimulus to counterregulation through the release of systemic and central peptides. It is likely that these mechanisms coexist because the stress response is at once essential to the survival of the species, and on the other hand, potentially highly toxic if sustained. It is therefore highly regulated at both the whole-body and cellular level.
Recurrent Hypoglycemia and Hypothalamic Glucose Sensing
Repeated hypoglycemia produces a downregulation of the hormonal counterregulatory response to subsequent hypoglycemia,[14] while its strict avoidance can restore the response.[42] Rodent studies indicate that changes in key brain glucose-sensing regions play a major role in mediating this phenomenon. Recurrent hypoglycemia markedly suppressed the counterregulatory response induced by local VMH perfusion with 2-deoxyglucose[43] and lowered the glucose level activating individual VMH glucose-sensing neurons.[44]
This adaptation might result from increased transport of glucose and/or alternate fuel into the sensing neuron. Repeated hypoglycemia increases the expression of glucose transporters at the blood-brain barrier[45] and increases whole-brain glucose uptake[46] and the uptake of the monocarboxylic acid acetate.[47] The effect on overall brain glucose transport has not, however, been observed in all studies,[48] raising the possibility that there is regional variation in the brain of this response. Alternatively, the central glucose-sensing neurons might obtain additional metabolic substrates from more local sources such as brain glycogen. Brain glycogen levels were reported to increase following the restoration of normoglycemia,[49] and this "super-compensation" could provide an additional fuel reserve. However, brain glycogen levels are very low (by necessity of the skull vault). In rodents, these levels also return to baseline within several hours of a hypoglycemic episode, a time when glucose counterregulation is still suppressed.[50] This does not, however, exclude the possibility of accelerated astrocytic glycogen turnover and in turn increased delivery of lactate following repeated hypoglycemia. In addition, repeated activation of the AMPK cascade would be expected to induce mitochondrial biogenesis and increased metabolism of fatty acids.[51] This potentially reduces neuronal demands for glucose, sparing it for other tasks. Interestingly, a recent study[52] comparing nondiabetic subjects with type 1 diabetic subjects who were unaware of their hypoglycemia found no difference in the overall rate of brain oxidative phosphorylation measured by 13C nuclear magnetic resonance, although the study was undertaken under euglycemic conditions.
As described earlier, acute hypoglycemia also activates a number of pathways involved in the regulation of the neuroendocrine stress response. Glucocorticoids,[38] CRH,[39] and urocortin 3[40] given under controlled euglycemic conditions (i.e., excluding hypoglycemia as a factor) can all induce defective counterregulation to next-day hypoglycemia. Activation of this family of neuropeptides plays an integral role in a number of different forms of stress, and they are tightly regulated. Studies in transgenic mice show that activation of CRH-R2 suppresses—whereas activation of CRH-R1 amplifies—the responses to a number of physiological stressors.[53] Therefore, an alteration in the balance between CRH-R2– and CRH-R1–mediated actions, induced by glucocorticoids or the CRH neuropeptides, could lead to suppression of the glucose counterregulatory response during a subsequent exposure to hypoglycemia. This mechanism would explain why the depth and duration of hypoglycemia both contribute directly to the magnitude of the subsequent counterregulatory defect (increased antecedent stress response),[54] and why alternate stressors, such as exercise,[55] induce similar changes.
These adaptations are not necessarily mutually exclusive and, given the complexity of the neuroendocrine response to hypoglycemia, it is likely that a number of adaptations at the cellular and whole systems levels all contribute to some degree in the development of defective counterregulation. In the authors' opinion, hypoglycemia initiates two primary adaptive responses, both of which are interlinked at many levels. The first results from hypoglycemia acting as an acute "starvation" signal leading to local cellular adaptations in the brain, such as an increased ability to use alternate fuels and changes to peripheral metabolism that would permit increased delivery of fuel substrates to the liver for the generation of glucose and ketone bodies. The second adaptation is a down-regulation of the stress response, which again takes place at both the cellular and whole systems levels and is designed to limit the potential of hypoglycemia to induce cell death. This later response is a very well-established response to repeated cellular stress and can be seen as a form of preconditioning or tolerance. These two principal adaptive responses also explain why there is likely to be regional variation in the effects of recurrent hypoglycemia. Neurons most affected by acute hypoglycemia (e.g., glucose-sensing neurons that are activated by hypoglycemia and drive the stress response) may show an enhanced dual effect of metabolic adaptation and feedback inhibition of the stress response caused by repeated hypoglycemia.
It is our belief that these changes are adaptive and not maladaptive and, to that extent, this would not be consistent with the current description of this phenomenon as hypoglycemia-associated autonomic failure. At a more basic level, repeated hypoglycemia is inducing hypoglycemia tolerance through preconditioning. This does not mean the individual is fully protected from the consequences of hypoglycemia. The problem is, of course, that the appearance of hypoglycemia in diabetes occurs when there is a marked hyper- rather than hypoinsulinemia. Hyperinsulinemia blocks peripheral generation of alternate fuels and, in the presence of impaired counterregulation, is more likely to induce severe and prolonged hypoglycemia. Under these conditions, brain extracellular fluid glucose levels are extremely low and, thus, there is the potential for cellular damage or even death. This is why the inability to exert feedback inhibition of insulin release and action during hypoglycemia is one of the key counterregulatory defects of type 1 diabetes.
Summary
Hypoglycemia remains a major obstacle to improved glycemic control in diabetes and, despite the development of novel short- and long-acting and insulin analogues and the more widespread use of pump therapy, the frequency of hypoglycemia in type 1 diabetes has not changed dramatically over the last 20 years. The challenge is to try and understand the mechanisms through which the body detects falling glucose and initiates a glucose counterregulatory response. Despite a few decades of research, these mechanisms are still poorly understood, as are the pathways through which different glucose-sensing regions communicate in order to integrate the whole-body response to hypoglycemia at a behavioral and a physiological level. A further challenge, but one that is crucial to the translation of this basic research into clinical practice, will be to examine candidate mechanisms in model systems that are more directly relevant to type 1 diabetes. There are currently no widely available therapies for the individual with type 1 diabetes who experiences recurrent severe hypoglycemia, so the need to develop such interventions is great.
Acknowledgments
The research work of the authors is supported by research grants from the National Institute of Diabetes and Digestive and Kidney Diseases (69831, 20495, and 45735), the Juvenile Diabetes Research Foundation, and the American Diabetes Association.
No potential conflicts of interest relevant to this article were reported.
R.J.M. and R.S.S. wrote and edited the manuscript.
The authors thank the postdoctoral fellows and technical staff who contributed greatly to the research that underpins this review.
Diabetes. 2010;59(10):2333-2339. © 2010 American Diabetes Association, Inc.
Cite this: Hypoglycemia in Type 1 Diabetes - Medscape - Oct 01, 2010.




Comments