Methods
The design and methodology for both studies were identical and are presented separately and combined, in accordance with guidelines suggested by the US Food and Drug Administration.
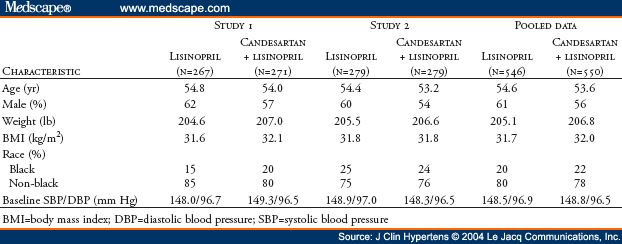
Eligible patients consisted of men and women >18 years of age with essential hypertension, characterized by a mean sitting diastolic blood pressure (DBP) of 90 mm Hg-114 mm Hg, inclusive, despite receiving treatment with lisinopril 20 mg daily for >4 weeks. Women of childbearing potential were required to use an effective method of birth control throughout both studies. Patients were excluded if they had secondary hypertension; mean sitting DBP >115 mm Hg or systolic blood pressure (SBP) >200 mm Hg; angina pectoris requiring more than short-acting nitrates; hemodynamically significant valvular heart disease; coronary angioplasty within the previous 3 months; myocardial infarction; coronary bypass surgery, stroke or transient ischemic attack within the previous 6 months; history of drug or alcohol abuse within the previous 2 years; significant renal impairment (serum creatinine level >2.0 mg/dL or serum potassium level >5.0 mEq/L); significant hepatic impairment; known hypersensitivity to ARBs or ACE inhibitors; current treatment with a dosage of lisinopril >20 mg daily; or current or prior treatment with candesartan, whether alone or in combination with other medications. All patients provided written informed consent before participating in the study.
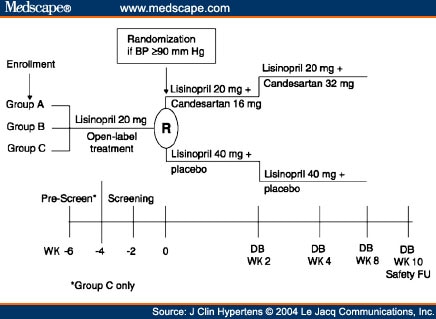
Local Institutional Review Boards approved the protocols for both studies. After a screening period in which patients received open-label lisinopril, patients whose BP remained uncontrolled (sitting DBP >90 mm Hg) and who satisfied the eligibility criteria entered an 8-week, double-blind phase during which they were randomized (1:1) to receive lisinopril (40 mg) alone or candesartan (16 mg increased to 32 mg) plus lisinopril (20 mg) (Figure 1). No other antihypertensive medications were permitted. In the monotherapy arm, patients received lisinopril 40 mg daily for the entire 8 weeks. Doses of lisinopril above 40 mg daily provide little additional antihypertensive activity.[16] In the combination arm, patients received candesartan 16 mg daily plus lisinopril 20 mg daily for the first 2 weeks and candesartan 32 mg daily plus lisinopril 20 mg daily for the remaining 6 weeks. The 6 weeks of treatment with candesartan 32 mg daily was expected to be sufficient to achieve the full antihypertensive effect of this agent. Doses of candesartan above 32 mg daily provide little additional antihypertensive effect.[17,18] Three groups of patients entered the initial open-label screening period (Figure 1):
|

Study schematic for Studies 1 and 2. Group A patients were receiving monotherapy with lisinopril 20 mg once daily for ≥ 2 weeks before study entry; Group B patients had newly diagnosed hypertension or hypertension untreated for ≥ 30 days before study entry; Group C patients were uncontrolled or intolerant to their antihypertensive treatment before study entry. BP=blood pressure; DB=double-blind; FU=follow-up; WK=week.
Patients visited the clinic for clinical evaluations every 2 weeks during the open-label screening period and at Weeks 1, 2, 4, and 8 (or premature withdrawal) during the double-blind phase. In addition, patients were contacted by telephone 2 weeks after the last dose of study medication for safety follow-up information. The double-blind study drug supplies were packaged in high-density polyethylene bottles using a double-dummy design. Placebo tablets were used to maintain blinding. Drug supplies were packaged to appear indistinguishable between study groups.
Lisinopril is an orally active, nonsulfhydryl ACE inhibitor that is widely used for hypertension.[16] Candesartan is a selective ARB devoid of agonist activity.[17] Insurmountable AT1 receptor blockade and long duration of activity result from distinctive AT1 receptor binding properties, such as high affinity and slow receptor dissociation rate.
At each clinic visit, trough (24 ± 2 hours postdose) sitting BP measurements were performed 3 times at 2-minute intervals using standard office mercury sphygmomanometers. The mean of three sequential BP readings (<5-mm Hg difference between the highest and lowest value) served as the BP determination for the visit. Patients were instructed not to take their study medication on the day of the clinic visit until after trough sitting BP measurements were obtained.
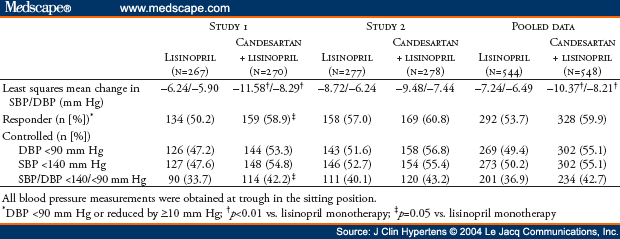
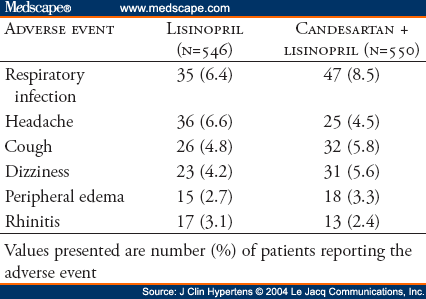
The primary efficacy measure of the antihypertensive effect was the mean change in trough sitting DBP from baseline to Week 8 of double-blind therapy. Secondary efficacy measures of the antihypertensive effect included mean change in trough sitting SBP, the proportion of responders at Week 8 (trough sitting DBP <90 mm Hg or reduced by >10 mm Hg), and the proportion of controlled patients at Week 8 (trough sitting DBP <90 mm Hg and trough sitting SBP <140 mm Hg). Safety was evaluated by monitoring of adverse events, standard laboratory tests (serum chemistry, hematology, and urinalysis), physical examinations, and heart rate.
Each study planned to enroll 494 patients to ensure 370 patients completed the study, assuming an alpha of 0.05, power of 85%, and a desired detectable difference in DBP of 2.5 mm Hg with a standard deviation of 8 mm Hg (two-tailed test). For the efficacy analyses, which are presented separately by study, the intent-to-treat (ITT) population included all randomized patients who received at least one dose of study medication and who had a baseline and at least one trough sitting DBP measurement during the double-blind phase of the study. A last observation carried forward (LOCF) approach was used to impute the Week 8 values for patients who withdrew from the study before Week 8. In addition, the analyses were repeated using actual data (no carrying forward of observations). Changes in trough sitting DBP and SBP from baseline to Week 8 were analyzed by analysis of covariance (ANCOVA) with the baseline BP as the covariate, and the changes were compared using least squares means from the ANCOVA model. Ninety-five percent confidence intervals (CIs) for the least squares mean changes from baseline and for the difference between the least squares mean changes from baseline were also calculated. Differences between the two treatment groups in rates and proportions (response and control rates) were compared by Fisher's exact test. Changes in trough sitting DBP and SBP were also analyzed by subgroups, based on race (black vs. non-black), age (>65 years vs. <65 years), gender, and diagnosis of diabetes using the ITT population.
The safety databases were pooled for presentation. All randomized patients who received at least one dose of study medication and who had at least one postbaseline contact with the investigational site were included in safety analyses. Although designed as two independent studies, the AMAZE program statistical analysis plans prespecified additional pooled analyses for patients with diabetes. A post hoc analysis was also conducted using BP data pooling from the two studies together using two-way ANCOVA methods with factors treatment, study, and the interaction terms in the model.
© 2004 Le Jacq Communications, Inc.
Cite this: Antihypertensive Efficacy of Candesartan-Lisinopril in Combination vs. Up-Titration of Lisinopril: The AMAZE Trials - Medscape - Sep 01, 2004.