Classification and Prevalence
The major biochemical defects for which the molecular background have been only partially elucidated are best classified by the main site at which the biochemical block occurs ( Table 1 ). Results from neonatal thyroid screening programs in more than 18 countries have indicated an overall incidence of CH worldwide of approximately, 1:4000 neonates. Females are affected twice as often as males. African Americans in the United States, have a lower incidence (1:10,000) than Asians and whites, the highest frequency being found in Hispanics.[9] An increased incidence has been noted in infants with Down syndrome.[10] Approximately 15%-20% of these have goitrous hypothyroidism, suggesting the presence of dyshormonogenesis. Therefore, the prevalence of biosynthetic defects in thyroid hormonogenesis is roughly 1:25,000 neonates.[11] The most common cause of CH is thyroid dysgenesis, which represents 80%-85% of all cases of permanent CH. CH secondary to pituitary or hypothalamic disease is the least common cause, occurring in just 1:50,000 to 1:100,000 infants. The incidence of the syndromes of resistance to thyroid hormone is unknown; a limited survey suggested the occurrence of 1 case in 50,000 live births.[12] More than 600 cases have been reported, of which 349 have been described in detail.[13,14] The incidence of complete TBG deficiency is approximately 1:15,000 newborn males.[15,16] Partial familial TBG deficiency has an average prevalence of 1:4,000 newborns, without racial or ethnic differences. The condition could be more common if TBG variants characteristic of distinct ethnic groups (such as Australian Aboriginal population) were included.
Genetic and clinical features of the syndromes of resistance to thyroid hormone have been reviewed recently[13,17] and this rapidly evolving field is outside the scope of this review (summarized in Table 1 ).
Hypothalamic-Pituitary Dysgenesis. Congenital central hypothyroidism might be caused by a dysfunctioning hypothalamus, pituitary gland or both caused by abnormalities in their ontogeny or a failure in the mechanism of thyrotropin-releasing hormone (TRH) and thyrotropin (TSH) synthesis and action.
The pituitary gland is formed by the fusion of two invaginations, one originating from the floor of the third ventricle and the other from the ceiling of the oral ectoderm (Rathke's pouch). A family of homeobox genes encode homeodomain transcription factors that control pituitary development. Genes coding for LHX3 (LIM homeobox protein 3), HESX1 (homeobox [expressed in embryonic stem cells] 1), and TITF1 (thyroid transcription factor-1) are essential for normal development of Rathke's pouch.[18] Other transcription factors, as PROP1 (prophet of Pit1, paired-like homeodomain transcription factor) and POU1F1 (POU domain, class 1) or Pit1 (POU domain, class 1, transcription factor 1) are required for the differentiation of the progenitor cells into differentiated cell lineages. These two transcription factors function sequentially in the same pathway to stimulate differentiation and proliferation of thyrotropic, lactotropic, and somatotropic cells.
Defects in LHX3. In the mouse, Lhx3 (human official gene symbol: LHX3) gene encodes for a transcription factor containing DNA-binding homeodomain and LIM domain that mediate protein/protein interaction and transactivation functions. The LIM proteins (named from the Lin-11, Isl-1, and Mec-3 genes that encode LIM/homeodomain family of transcription factors) are involved in the control of cell lineage determination and the regulation of differentiation. Lhx3 is expressed in the embryonic rodent brain and spinal cord and later is restricted to the developing and adult pituitary gland. Disruption of both alleles in the mouse Lhx3 -/- locus results in depleted thyrotrophs, somatrotophs, gonadotrophs, and lactotrophs, while corticotrophs fail to proliferate, demonstrating that Lhx3 is important to pituitary development fate commitment.
The human homologue gene LHX3 maps to chromosome 9q34.2-34.3, comprises 7 exons and spans 7.2 kb.[19]
Homozygous LHX3 mutations have been described in two unrelated consanguineous families. The affected family members presented with a combined complete deficit in all but one (adrenocorticotropin) anterior pituitary hormones, retarded growth, and a rigid cervical spine leading to limited head rotation. The LHX3 mutations consist of a missense mutation (Y116C) in the encoded LIM protein and an intragenic deletion that results in a truncated protein lacking the DNA-binding homeodomain.[20] It was shown that the tyrosine missense mutation inhibits the ability of LHX3 to induce transcription from selected target genes but does not prevent DNA binding and interaction of LHX3 with partner proteins. Mutant LHX3 missing a homeodomain does not bind DNA and is not able to induce transcription from pituitary genes.[21]
Defects in HESX1. In mice, Hesx1 (human official gene symbol: Hesx1) encodes a protein regarded as the earliest transcription expressed within the pituitary primordium and it likely influences normal development of forebrain, eyes, hypothalamus, pituitary gland, and the olfactory bulbs. Defects in the mouse gene Hesx1 and human Hesx1 are associated with septo-optic dysplasia in mouse and humans, respectively. This entity is characterized by hypoplasia of the optic nerve, various types of forebrain defects, and pituitary hormone deficiencies including hypothyroidism.
A study of 38 patients with septo-optic dysplasia showed that two siblings from a consanguineous family presented with panhypopituitarism, absent septum pellucidum, optic nerve hypoplasia, and agenesis of the corpus callosum. These subjects harbored a homozygous R53C missense mutation in a highly conserved amino acid in the Hesx1 homeodomain that affected the protein ability to bind DNA.[22] Heterozygous family members were phenotypically normal and no mutations of Hesx1 were found in 18 sporadic cases of optic nerve dysplasia. In 228 patients with congenital pituitary defects, ranging in severity from isolated growth hormone to septo-optic dysplasia with panhypopituitarism only 3 heterozygous Hesx1 missense mutations were found.[23] Two brothers were shown to present a S170L mutation associated with growth hormone (GH) deficiency and in one of them optic nerve hypoplasia. Another patient with isolated GH deficiency showed a T181A mutation. A G18C mutation was identified in a patient with multiple anterior pituitary hormone deficiencies.
Defects in PROP1. PROP1 gene encodes a paired-like homeodomain protein that is expressed specifically in the embryonic pituitary gland and is necessary for Pit1 gene expression. Alteration in this gene results in an apparent failure of initial determination of Pit1 lineage required for the production of GH prolactin (PRL), and TSH. The human PROP1 gene is located on the distal end of chromosome 5q.[24] PROP1 gene mutations have been observed in the AMES dwarf mouse, sporadic and familial human cases. Homozygous or compound heterozygous inactivating mutations were identified in the affected family members.[25] This study established that defects in PROP1 are responsible for subtypes of the multiple pituitary hormone deficiency syndrome that includes GH, TSH, PRL, follicle-stimulating hormone (FSH), and, luteinizing hormone (LH) deficiency. A 2-bp deletion in the AG-rich region of exon 2 in the PROP1 gene appears to be a hot spot[26] as was confirmed in a study describing eight affected patients homozygous for this mutation.[27] The phenotype observed in these patients is compatible with the complete loss of PROP1 activity. All patients (hormonal therapy stopped) had short stature and were sexually immature; their intelligence was normal, probably because the hypothyroidism was considered to be mild.[27,28] Three of them presented with an increased sella turcica area documented by lateral skull films. The degree of sellar enlargement varied in these patients. The hormone deficiency phenotype of affected individuals vary according to the onset of hormone deficiencies at different time points. Also, patients with the same mutations may differ in terms of hormone deficiencies.
Defects in POU1F1. The differentiation and proliferation of three pituitary cell lines (somatotrophs, lactotrophs, and a subset of thyrotrophs: TSH-β) was shown to coincide with the onset of expression of a unique pituitary specific transcription activating factor (mice: Pit1; human official gene symbol: POU1F1), which binds to enhancer elements in the flanking regions of the GH, PRL, and TSH genes.[29] Aside from its role in pituitary development, the transcription factor POU1F1 can act as the main stimulator for GH, PRL, and TSH synthesis binding to cis-acting elements of the promoters of these genes.[29] Combined pituitary hormone deficiencies of TSH, GH, and PRL have been described in patients with congenital hypothyroidism.[30,31,32,33,34,35,36,37] Consanguinity was present in some of these kindreds. Provocative testing has indicated low or absent levels of TSH, GH, and PRL but normal LH, FSH, and adrenocorticotropic (ACTH) in response, respectively, to luteinizing hormone-releasing hormone (LH-RH) and metyrapone.
Polymerase chain reaction (PCR) was used to amplify each of the six exons of POU1F1. POU1F1 gene mutations have been found in eight unrelated patients with various clinical presentations.[32,33,34,35] More recently a mother-infant pair was found to be heterozygotic for a point mutation in codon 271 of the gene encoding POU1F1, resulting in fetal hypothyroidism with neurologic damage.[37] Both sporadic and familial forms of POU1F1 gene mutations have been reported and the codon 271 is a common site for these mutations. In most cases imaging of the pituitary reveals pituitary hypoplasia. Serum GH and PRL levels are usually immeasurable but serum TSH and thyroid hormone levels are variable. At a later age, the GH deficiency usually dominates the clinical manifestations. Central thyroid deficiency may develop only after GH replacement therapy has begun. The clinical heterogeneity and inheritance patterns of these patients suggest that POU1F1 gene mutations are a more frequent cause of anterior pituitary hormone deficiency than previously recognized. Depending on the location of the mutation, one mutated allele is sufficient to result in an abnormal phenotype.
TSH is synthesized in the pituitary gland and is regulated by both TRH and the thyroid hormone negative feedback system (Fig. 1).
The hypothalamic-pituitary-thyroid axis and key steps in thyroid hormone formation. Hormone synthesis requires active transport of iodide across the basolateral membrane by sodium iodide symporter (NIS). At the apical membrane, pendrin and human apical transporter of iodide (hAIT) will transport iodide to the colloid, thyroid peroxidase (TPO) organifies iodide into tyrosine residues on Tg generating monoiodotyrosine (MIT) and diiodotyrosine (DIT). MIT and DIT are coupled to triiodothyronine (T3) and thyroxine (T4). Both steps require the presence of H2O2. After pinocytosis, T3 and T4 are secreted into bloodstream. MIT and DIT are deiodinated by a dehalogenase. TRH, thyrotropin-releasing hormone; T3H, thyrotropin.
Defects in TRH Receptor. The TRH receptor (human official gene symbol TRHR) belongs to the superfamily of G-protein coupled receptors. These receptors have a conserved structure that is characterized by an extracellular N-terminal domain, 7 hydrophobic helices inserted into the membrane, 3 intracellular and 3 extracellular loops, and an intracellular C-terminal domain.[38] The activated TRH receptor is mainly coupled via regulatory G proteins to the phosphoinositide-protein kinase C pathways. Central congenital hypothyroidism with complete absence of TSH and PRL responses to TRH has been described in a 9-year-old boy.[39] This patient presented mild symptoms of hypothyroidism, short stature with markedly delayed bone maturation. Two brothers and the parents were clinically euthyroid. The parents and the oldest brother were heterozygous for the defect and phenotypically normal. The patient was compound heterozygous for two mutations in the coding region of the gene. In vitro studies on the expressed mutated TRHR indicated that both mutations resulted in a biologically inactive receptor, unable to bind TRH.
Defects in TSH-beta Subunit. TSH (human official gene symbol, TSH) stimulates the function and growth of the thyroid via interaction with a specific plasma membrane receptor. TSH consists of two different (
 and β) noncovalently linked subunits. Whereas the
-subunits of the glycoproteins LH, FSH, and human chorionic gonadotropin (HCG) are identical, the β-subunits are unique for each of these hormones and carry specific information for receptor binding and hormonal action. The TSH-β gene is located on chromosome 1p22.[40] For biologic activity heterodimerization is required.[41] Congenital hypothyroidism because of a lack of TSH synthesis and to TSH with impaired biologic activity have been reported.
and β) noncovalently linked subunits. Whereas the
-subunits of the glycoproteins LH, FSH, and human chorionic gonadotropin (HCG) are identical, the β-subunits are unique for each of these hormones and carry specific information for receptor binding and hormonal action. The TSH-β gene is located on chromosome 1p22.[40] For biologic activity heterodimerization is required.[41] Congenital hypothyroidism because of a lack of TSH synthesis and to TSH with impaired biologic activity have been reported.
In some cases an increased serum concentration of aberrant TSH can compensate for the impaired biologic activity. Other patients showed undetectable serum TSH concentrations but normal concentrations of other pituitary glycoprotein hormones. These are autosomal recessive defects causing typical symptoms of congenital hypothyroidism such as mental and growth retardation. At least 24 patients have been described in 16 unrelated families.[42,43,44,45,46,47,48,49,50,51] The patients were clinically hypothyroid, with low serum thyroid hormone levels, low iodine uptake that increased after administration of exogenous bovine TSH, and low or undetectable serum concentrations of TSH. After stimulation with TRH both serum TSH and free TSH-β subunit were consistently undetectable. By contrast, the levels of serum free TSH-
were high and significantly increased after TRH administration. The elevated levels of TSH-
subunit decreased to an undetectable level during replacement therapy with triiodothyronine, indicating that the thyrotrophs were responsive to the suppressive effect of thyroid hormone.
Both conventional insulin and LH-RH stimulation tests revealed normal increases of serum GH, LH, FSH, and ACTH. Thus the defect is restricted to the TSH-β subunit gene. In two kindreds, molecular biologic studies have indicated mutations in two different sites of exon 2, generating a peptide that would dimerize with subunits to synthesize TSH molecules. In one kindred a truncated TSH-β protein was translated that generated a biologically inactive but detectable serum TSH molecule.[44] The mutation found in four Japanese is G29R that altered the β-polypeptide, making it unable to be associated with the
-subunit.[44] Moreover, the mutation found by the authors[46] and several other groups results in a frameshift with a longer polypeptide. Thus not all TSH-β mutations result in premature stops. The mutated TSH-β protein cannot associate with the
-subunit to produce a functional TSH heterodimer. As a consequence, an excess of
-subunits is released into the circulation with a hyperplastic pituitary gland.[42,43,44,45] In a Brazilian family some
-subunit combination occurred with the mutant TSH-β subunit molecule.[46] The authors were able to demonstrate that when synthesized by a mammalian expression system transfected with the wild-type and mutant TSH-β genes the heterodimeric TSH molecule had reduced biologic activity. In another recent study, Pohlenz et al.[52] were able to demonstrate a mutation at position +5 of the donor splicing site, producing a skip of exon 2. A severely truncated peptide of only 25 amino acids was translated ( Table 2 ).
Defects in TSH Receptor. The TSH receptor (human gene official symbol, TSHR) is a member of the large family of G-protein-coupled seven transmembrane domain receptors. The long amino-terminal domain (extracellular) encodes the specificity for thyrotropin recognition and binding. Binding of TSH stimulates the exchange of GDP (guanosine diphosphate) for GTP (guanosine triphosphate) on the
-subunit (G
) of the trimeric G protein (G
, β,
 ). After dissociation of the trimer, the
-subunit interacts with downstream effectors of the receptor.
). After dissociation of the trimer, the
-subunit interacts with downstream effectors of the receptor.
In the case of the TSH receptor, as reviewed recently,[53] the main G-protein involved is Gs which activates adenylyl cyclase via Gs
. TSH is also active through a second messenger system cyclic adenosine monophosphate (cAMP)-independent (the PIP2 pathway) where the G-protein involved is Gq. As expected, the cytoplasmic concentrations of cAMP control the level of functional activity of the thyroid gland and the expression of thyroid-specific genes such as thyroglobulin, thyroperoxidase, and the TSHR itself. Both excessive or unregulated thyroid function and lack of responsiveness to TSH causing hypothyroidism has been described caused by mutations of the TSHR gene. Hyporesponsiveness to TSH may occur because abnormalities in the TSH molecule or in the TSH stimulatory pathway.
For a hormone receptor, gain-of-function may have three meanings: activation in the absence of ligand (constitutivity), increased sensitivity to its normal agonist, or broadening of its specificity.[54] When the receptor is part of a modulatory mechanism, the first situation may lead to tissue autonomy, whereas the second would be expected to result in reduced concentration of the agonist. In the third case, inappropriate stimulation of the gland is expected to occur because the indiscriminate agonist would not be subjected to the normal negative feedback. If a gain-of-function mutation of the first category occurs in a single cell expressing the receptor (somatic mutation) normally, it only will become symptomatic if the regulatory cascade controlled by the receptor is activated in this particular cell type. Autonomous activity of the receptor will cause clonal expansion of the mutated cell. If the regulatory cascade also controls positive function, the resulting tumor will progressively take over the function of the normal tissue, ultimately leading to autonomous hyperfunction (Fig. 2). If the mutation is present in all cells of an organism (germline mutation), autonomy will be displayed by the whole tissue expressing the receptor. In case where the regulatory cascade is both mitogenic and activates function, the expected result is hyperplasia associated with hyperfunction.[54] Loss-of-function mutations in the TSHR gene are expected to cause a syndrome of resistance to TSH or unresponsiveness of the follicular cell to TSH. The expected phenotype is likely to resemble that of patients with mutations in TSH itself.[54]

Mutations of the thyrotropin (TSH) receptor causing hyperthyroidism (activating mutations) or hypothyroidism (inactivating mutations). TG, thyroglobulin; T4, thyroxine. Modified from Guillam and Kopp.[5]
Loss-of-Function Mutation. Resistance to TSH may be caused by various molecular mechanisms.[53] In a subset of patients, the molecular cause consists of inactivating mutations in TSHR that are partially or completely inactivating. The mode of inheritance is recessive and affected individuals are homozygous or compound heterozygous for the mutations. Among these patients the phenotype encompasses a wide spectrum ranging from isolated TSH elevation to severe hypothyroidism and there is a clear correlation between genotype and phenotype. In other patients with sporadic or familial resistance to TSH, the TSHR gene was found to be normal, indicating locus heterogeneity caused by to defects in other genes.[53] From an historical point of view, this syndrome was described almost 30 years ago,[55] and hypothetically attributed to a defective action of TSH.[55,56,57] The first patient with suspected impaired thyroid response to thyrotropin was a 8-year-old male child, severely retarded and hypothyroid. He had a normally positioned thyroid that did not respond adequately to his own elevated serum level of biologically active TSH.[55] Other patients[56,57,58] were subsequently described and in vitro studies confirmed that there was normal binding of TSH to the TSHR. However the TSH stimulation of the TSHR-adenylate cyclase system was markedly decreased, resulting in decreased hormone synthesis and thyroid cell growth. Three siblings with congenital hypothyroidism were found to be unresponsive to TSH stimulation.[59] Sequencing their (cDNA)TSHR failed to indicate a significant mutation in the TSHR gene. In another family, reported by Sunthornthepvarakul et al.[60] three sisters, offspring of unrelated parents, were found to be euthyroid but displayed elevated TSH concentrations in their plasma ( Table 3 ). Thyroid hormone serum levels were normal (compensated hypothyroidism). The three affected siblings were found to be compound heterozygous for mutations in the extracellular TSH binding domain of the receptor (P162A and I167N). In vitro studies documented that the I167N mutation had almost no biologic activity, whereas P162A displayed reduced activity.[60] When tested in transfected COS cells, the maternal allele displayed a loss of sensitivity to TSH and the paternal allele was almost completely nonfunctional. Clearly the affected siblings of this family had only a mild clinical disturbance of the thyroid function, unlike the other reported cases. Possibly different mutations cause more complete inactivation of the TSHR in cases of severe congenital hypothyroidism.
To date, at least 11 pedigrees with a total of 15 different mutations have been reported.[53] Of 15 loss-of-function mutations in the TSHR gene, 9 mutations were in the extracellular domain and 6 mutations in the transmembrane segments. These mutations were shown to affect ligand binding, signal transduction, and/or cell surface expression of the receptor.
More recently, four unrelated families with loss of function mutations in the TSHR gene were described.[61] One patient has a homozygous P162A substitution. The others were compound heterozygotes. Expression of the various mutated receptors in transfected COS-7 cells demonstrated the impairment of their function. Two novel mutations in the TSHR gene were recently described in a child with resistance to TSH.[62] One allele has a G > A transition corresponding to an arginine to glutamine change at codon 109 (R109Q) in the extracellular domain and the other allele has a G > A transition corresponding to a premature termination codon at tryptophan 546 (W546X) in the fourth transmembrane segment.[62] Very recently, two novel inactivating missense mutations at codons 450 (R450H) and 498 (G498S) located in the first cytoplasmic loop and third transmembrane domain, respectively were reported in Japanese children.[63]
Two animal models have been described with thyrotropin resistance: mutant cats (dfc/dfc)[64] and the homozygotic (hyt/hyt) mouse.[65] Both species have nongoitrous primary hypothyroidism and elevated serum TSH. A mutation in the TSHR was found in hyt/hyt mice,[65] located in the fourth transmembrane segment. The molecular defect in the dfc/dfc cat is not known.
It is interesting to analyze the location of these mutations and the relative loss of function of the TSHR in the light of a recent structural model for the extracellular domain of the TSHR.[53,60,61,62,63] It was proposed that the inside concave surface of the leucine-rich repeats of the amino terminal portion of the TSHR, composed of beta sheets, would provide the recognition surface for TSH binding.
It is possible that other clinical entities such as congenital hypothyroidism with thyroid hypoplasia (the thyroid gland in the normal cervical position) or athyreosis may also have a defective interaction of the endogenous TSH with its receptor. An apparent congenital athyreosis[66] in a 2-week-old boy with high serum TSH levels was found to be harboring a splicing mutation in the maternal allele (G > C transversion at position +3 of the donor site of intron 6) and the other allele a deletion of 2 bases in codon 655 of exon 10 (compound heterozygosity). The patient had undetectable uptake on 99mTc-pertechnetate scintigraphy (no visible thyroid tissue) but normal serum thyroglobulin, thus demonstrating an ectopic or hypoplastic thyroid gland, indicating that ultrasound is preferred as the first-line imaging study because the lack of sensitivity of scanning. This finding show that inactivating mutations in TSHR in hypothyroid newborns may account for some cases of the so-called apparent congenital athyreosis and should be suspected, especially if serum thyroglobulin concentrations are measurable.
Resistance to TSH without Mutations in the TSH Receptor. As mentioned, Takeshita et al.[59] analyzed the nucleotide sequence of the entire coding region of the TSH receptor gene in three patients with primary congenital hypothyroidism caused by TSH unresponsiveness who were the offspring of consanguineous parents. The TSHR was sequenced by a reverse transcription-polymerase chain reaction (RT-PCR) procedure using RNA extracted from leukocytes and did not show abnormalities. One patient was found to be heterozygous for a known polymorphism (Y601H).
Linkage studies using genetic markers flanking the TSH receptor by a distance of 2.3 and 1.2 cMo were performed in 23 cases with congenital hypothyroidism with familial occurrence of consanguineous parents by Ahlbom et al.[67] These analyses did not support linkage to the TSHR locus.
In another study of four unrelated children with congenital hypothyroidism and eutopic hypoplastic gland, Nogueira et al.[68] were unable to find defects in the coding region of the TSHR.
Similar observations was mentioned by Xie et al.,[69] who reported a study of three unrelated families with resistance to TSH without abnormalities in the coding region or promoter of the TSHR gene.
These aforementioned studies outline that TSH resistance is genetically heterogeneous and may result from other molecular defects than TSHR mutations.[70]
Somatic Mutations in Autonomous Thyroid Nodule. The concept that mutations in the TSHR could constitutively activate adenylyl cyclase in the thyroid came from studies with other receptors such as the adrenergic
1b receptor, the MSH receptor gene and the rhodopsin gene.[71] Because solitary toxic thyroid adenomas are well-limited, encapsulated benign tumors characterized by autonomous function (TSH-independent), it was thought that long-term activation of the cAMP regulatory cascade might be present. In a series of 11 toxic adenomas a somatic mutation was identified in 9.[72] Five different residues were found to be affected by a total of six different amino acid substitutions (Fig. 2).
Other studies have added more mutations to this list.[73,74,75,76] Surprisingly, the frequency of constitutively activating TSHR mutations in a large series (n = 45) of autonomous nodules in Japan was remarkably low.[77] Only one hyperfunctioning adenoma displayed a 3-base deletion (nt 1953-1957). Also, the frequency of TSHR mutations was evaluated in 40 toxic thyroid adenomas in Italy.[78] Activating mutations were found in 25% of the examined tissues in codons 619, 631, 632, and 633. Mutations have been described in the first and second extracellular loops, the sixth transmembrane segment, and the second extracellular loops. Activating mutation of the TSHR gene (position 633) was also recently described in aggressive insular thyroid carcinoma, and in lymph node metastasis.[79] Presumably other parameters are involved in the clinical course of the disease, in addition to the nature of the mutations (such as iodine content of the diet, other gene loci involved in the thyroid physiology, the growth rate of the mutated clone). The possibility of multiple mutations in the TSH in toxic nodules of multinodular goiter has been considered possible.[53] The increase in cell number because of long-term stimulation and the generation of free radicals within the multiple nodules could be considered as inducing factors for mutations.
In a small series from Brazil, 6 of 7 autonomous thyroid adenomas harbored heterozygous mutations known to confer constitutive activity to TSHR.[80] These differences in frequency may indicate that other oncogenic mutations are present in toxic adenomas. A solitary toxic adenoma harboring a novel mutation (S281I) was found[81] in a newborn with clinical signs of hyperthyroidism shortly after birth. The identification of this mutation demonstrates that activating mutations also occur in the extracellular domain of the TSHR. Congenital hyperthyroidism due to a constitutively activating TSH receptor mutation has to be considered if neonatal hyperthyroidism is persistent, and parameters of autoimmunity are absent.
Germline Mutations in Hereditary Toxic Thyroid Hyperplasia. A pedigree in which hyperthyroidism (nonautoimmune) segregates as an autosomal dominant trait was described in France[82] with absence of ophthalmopathy or biologic signs of autoimmunity (repeatedly negative circulating autoantibodies such as anti-thyroid peroxidase [TPO], thyroid-stimulating or blocking antibodies). Also, there was no lymphocytic infiltration in the thyroid tissue removed at surgery. The goiters developed gradually and were of variable size. The age of onset of hyperthyroidism was highly variable, from 18 months to adulthood. Despite these differences in expression, a common characteristic of the cases is that aggressive treatment such as radical surgery or ablation by radioiodine seemed required to avoid relapse. By analogy with toxic thyroid adenomas this disease entity was appropriately termed toxic thyroid hyperplasia and similar mutations at the germline level were thought to be the cause of the syndrome.[82,83] Two different mutations of the TSHR were initially identified in two pedigrees from France and were localized in transmembrane segments 3 and 7. Their functional characteristics, after expression in COS cells, were similar to those already described in toxic adenomas, (e.g., stimulation of cAMP accumulation and no effect on the phospholipase C-dependent pathway[84]). Since the initial study, three other families, each with a different mutation, have been identified, which suggests that the condition may be more frequent than originally thought.[69] In addition, newborns with sporadic congenital hyperthyroidism were shown to harbor germline neomutations in the TSHR gene.[81,85] One mutation[85] was in a residue that had also been found mutated in toxic adenomas (position 631). In four other patients from two unrelated German families[85] an exchange of alanine for valine was found at the position 623 (A623V) and, an exchange of serine for asparagine at position 505 (S505B) of the TSHR.
Ontogeny of the Thyroid Gland. The thyroid gland is the first endocrine gland to appear in embryonic development. The gland develops from a median endodermal thickening in the floor of the primitive pharynx. This thickening, known as the thyroid diverticulum, moves caudally. By 7 weeks gestation, the human thyroid gland has usually reached its final site in the neck. During thyroid embryogenesis, genes encoding thyroid-specific transcription factors are expressed at the onset of gland formation and during gland migration. The transcription factors TITF1, FOXE1, and PAX8 are crucial for thyroid development[86] ( Table 4 ). Genes specific for the differentiated follicular cell (TSHR, TG, and TPO) are expressed later.
Defects in TITF1. The first gene to be expressed during thyroid formation is that encoding TITF1(thyroid transcription factor 1; previous symbol NKX2A; official gene symbol TITF1), formerly called TTF-1, essential for developmental processes. TITF1 is expressed in thyroid, lung, and several structures of forebrain. TITF1-deficient mice lack lungs, thyroid gland, the pituitary gland and show profound alterations in brain development.[88]
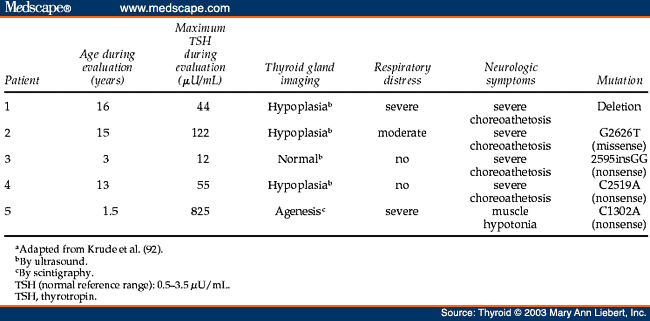
Although it appears that mutations in the TITF1 gene (maps to human chromosome 14q13) are not frequent,[89,90] a patient having a heterozygous deletion of chromosome 14q12-13.3 was congenitally hypothyroid and showed severe respiratory distress syndrome.[91] Furthermore, a recent study reported patients who showed a clinical picture of choreoathetosis, mental retardation, thyroid abnormalities, and pulmonary complications.[92] In these five patients with variable degrees of CH, Krude et al.[92] found heterozygous loss of function mutations in each of them: one complete gene deletion, one missense mutation (G2626T), and three nonsense mutations (2595-2596insGG, C2519A, C1302A) ( Table 5 ). The association of symptoms in the patients with TITF1 mutations indicates an important role of human TITF1 in the development and function of thyroid, basal ganglia, and lung ( Table 5 ), as demonstrated for genetically engineered animals in which one allele is fully inactivated.[93] This pattern of organ manifestations of TITF1 gene mutations appears to be similar in the affected patients ( Table 5 ). The clinical similarity in the patients affected by a large deletion of chromosome 14q and by loss of function mutations within TITF1 gene implies that this phenotype is entirely the result of TITF1 haploinsufficiency.[92,93] Human mutations often inactivate one allele, producing haploinsufficiency, or a reduction by half in the protein levels of the encoded transcription factor. As evident from a review of the plethora of clinical manifestations associated with genetic syndromes, half-normal levels of many transcription factors synthesized by the normal allele are not sufficient for protein function.[94] The prominent finding of nonprogressive choreoathetosis in TITF1-deficient patients may suggest that other congenital extrapyramidal movement disorders could be caused by TITF1 mutations. This is supported by recent link of benign hereditary chorea to a region on chromosome 14 including the TITF1 gene locus.[92]
Defects in FOXE1. FOXE1 (forkhead box E1 [thyroid transcription factor 2]) is a member of a large family of proteins that bind DNA through a sequence called the forkhead domain. The human gene (previously designated TITF2, FKHL15, or TTF-2; official gene symbol FOXE1) maps to chromosome 9q22.[95] This transcription factor is expressed in the thyroid gland, in most foregut endoderm, in the craniopharyngeal ectoderm, involved in palate formation, and also in Rathke's pouch. Mice lacking the Titf2 locus (Titf2-/-) die within 48 hours of birth, mostly because of severe cleft palate. These mice do not have a normally localized thyroid gland. The pituitary gland responded normally to the decreased free thyroxine (T4) serum levels by elevated serum TSH concentrations.[96]
A mutation in FOXE1 associated with CH, cleft palate, and choanal atresia has been described in two siblings with a homozygous missense mutation (A65V) within the forkhead domain of the gene.[97] The mutant protein has impaired DNA binding and loss of transcriptional function. In addition, in human patients spiky hair and a hypoplastic bifid epiglottis have been mentioned ( Table 4 ).
Defects in PAX8. The third transcription factor known to be essential for thyroid development is mice Pax8 (human gene official symbol, Pax8), which binds DNA via the conserved paired domain and is one of the transcriptional regulators that control differentiation.[98] The Pax8 gene maps to human chromosome 2q12-q14 and is expressed in the thyroid diverticulum, the developing mid- and hindbrain and the kidney.[99] In the thyroid, Pax8 in involved in thyroid development and TPO and TG gene expression.[100] In mice with disrupted Pax8 locus, the eutopic thyroid is smaller and has no follicular organization; the gland is composed almost completely of calcitonin producing C cells.
The Pax8-inactivating mutations reported in humans have all been found to be monoallelic mutations.[101] Two patients were found in a population of 145 children with CH. One, with a dystopic thyroid remnant, was heterozygous for an Arg108Stop transition (A108X). The other, with a dysgenetic eutopic thyroid, was heterozygous for an A31H mutation. Both transitions appeared to be de novo mutations. In addition to these two, another family in which CH occurred as a result of a dysgenetic eutopic thyroid has been described. This family also harbored a monoallelic L62A mutation. In vitro assays showed that the mutated Pax8 molecules were unable to bind to or activate a TPO promoter coupled to a luciferase gene. This shows that these mutations could cause a severe loss of Pax8 function as a consequence of decreased DNA binding. However, haploid insufficiency[94] is considered as a plausible explanation why one mutated Pax8 allele in humans leads to congenital hypothyroidism,[101,102] recent reported findings do not support it. A novel Pax8, Q40P mutation heterozygous transversion, found in a patient with CH and thyroid hypoplasia confirms the role of Pax8 in normal thyroid development.[103] However, the absence of thyroid hypoplasia and hypothyroidism in the mother of the affected index patient suggests that Pax8 mutations have a variable penetrance or expressivity.[103] Of particular interest is the variability in morphologic phenotype among patients with Pax8 mutations ( Table 6 ).
Associated Congenital Malformations and Thyroid Anomalies in Infants with CH. In the last decade a high frequency of other congenital anomalies have been reported in infants with CH detected by neonatal screening.[105] In Italy 5.5% of infants (n = 1420) with CH had cardiac anomalies (atrial septal defects, isolated pulmonary stenosis, tetralogy of Fallot).[106] Another 2.9% of the neonates had anomalies of the nervous system, eyes or multiple congenital malformations bringing the total to 8.4% of all infants with CH. This is fourfold higher than the reported frequency in the healthy Italian neonates (1-2%).[106] Isolated cardiac malfunctions (3.0%) were also observed in a Canadian group of infants with CH, a prevalence that was fivefold higher than that in the general population.[105] The cardiac malformations were largely due to atrial and ventricular septal defects ( Table 7 ).
More recently Leger et al.[116] reported a significantly higher presence of thyroid developmental anomalies (thyroglossal duct cysts, pyramidal lobe, thyroid hemiagenesis and ectopic thyroid tissue) among first-degree relatives (n = 241) of 84 isolated children with CH with thyroid dysgenesis. A segregation analysis led to the conclusion that thyroid developmental abnormalities are compatible with an auto-somal dominant mode of inheritance with a low penetrance.
Iodide Transport Defect. The first step in thyroidal iodine metabolism is the cellular uptake of iodide from the extracellular fluid (Fig. 3). The inborn error involving the transport of iodide into the thyroid and other tissues is a rare cause of thyroid dyshormonogenesis. The possibility of a defect in iodide transport was first brought forth with the elegantly presented study of a hypothyroid boy of a consanguineous marriage, reported in 1960 by Stanbury and Chapman.[117] This study also demonstrated that the defect in the iodide transport was not limited to the follicular cell, but also affected other iodide-concentrating tissues such as the salivary glands and the gastric mucosa. Later, Wolff[118] extended these observations by demonstrating that the normal ability of the choroid plexus to concentrate iodide was also absent.

Diagram of a thyroid cell showing its morphologic polarity and components of the iodide transport system. The ion influx involves an active accumulation across the basolateral membrane mediated by a glycoproteic carrier sodium iodide symporter [NIS] in an electrogenic process, with the Na+ gradient as the driving force, generated by the Na+/K+-AT-Pase. From the follicular cell the iodide moves across the apical membrane transported by pendrin and human apical iodide transporter (hAIT), possibly regulated by thyrotropin (TSH) via cyclic adenosine monophosphate (cAMP). The iodide is delivered to a cell-colloid interface, the site of peroxidase-catalized thyroid peroxidase [TPO] iodide organification. The colloid-stored iodide eflux, also TSH regulated, occurs in the opposite direction across the apical membrane toward the interstitium. ADP, adenosine diphosphate; ATP, adenosine triphosphate; T3, triiodothyronine; T4, thyroxine.
Iodide is concentrated against an electrical potential of 50 mV and under physiologic condition the thyroid iodine to serum (T/S) ratio is approximately 20-40/1.[119] The accumulation of iodine in the thyroid is mediated by the sodium iodide symporter (NIS), an intrinsic plasma membrane carrier protein located in the thyroid follicular cells.[120] The human NIS was cloned from a human thyroid cDNA library in l996.[121] The gene coding for human NIS has been mapped to chromosome 9p12-13.2, has 15 exons, coding for a glycoprotein of 643 amino acids. As a member of the sodium-dependent transporter family, NIS is an intrinsic membrane protein with 13 putative transmembrane domains, an extracellular amino terminus and an intracellular carboxyl-terminus[122] (Fig. 4). To date several cases have been reported worldwide.[123,124,125,126,127,128,129,130,131,132,133] The various NIS mutants found in patients with iodide transport defects have been shown to be inactive (Fig. 4). However, the precise structural change brought about by a NIS mutation has only been established for the recurrent T354P mutation.[124]

Structure of the human sodium iodide symporter (NIS) based on a model predicting a transmembrane domain of 13 segments. The location and nature of known mutations are indicated. Note the recently reported deletion (del 142-323) extending from exon 3 to exon 7. (Modified from Pohlenz and Refetoff[128]).
The affected subjects characteristically present with congenital goiter and hypothyroidism, a defect in the iodide trapping mechanism in the thyroid and salivary glands, as well as in the gastric mucosa and choroid plexus.[117,118,134,135] The parents frequently are consanguineous, and there is a familial incidence of goiter. As expected, all subjects exhibited high serum cholesterol levels, serum T4 less than 1.0 µg/dL and high serum TSH levels. However, although large multinodular goiters are found on physical examination, the radioiodine uptake is less than 5% at 2, 6, and 24 hours, and there is no further increase after exogenous TSH stimulation. Some patients reported with this error in thyroid function have a complete defect, with saliva/plasma ratios close to 1.0, whereas others had only partially impaired transport. The amount of iodine per gram of thyroid tissue is significantly higher in patients with the partial defect than in the complete form. The disease has been corrected by administration of iodine, suggesting that under favorable conditions enough iodide can enter the gland by diffusion to allow adequate production of thyroid hormone.[119] However, concentrated supplies of iodide are not available in nature and a severe limitation of hormone synthesis may be imposed by chronic iodide deficiency. This implies an intimate interaction between genetic potential and environment in determining the phenotype. The goiters described in this syndrome have been large and multinodular. Histologically they are intensely hyperplastic, with scant or absent colloid. Electron micrographs show rudimentary follicular lumina filled with long microvilli and dilatation of the rough endoplasmic reticulum.[135]
Defects in Iodide Organification and Coupling of Iodothyrosines. After iodide is transported into the thyroid cell it is oxidized and bound to tyrosyl residues present in thyroglobulin (TG). This step in thyroid hormonogenesis depends upon intact TPO activity, generation of an oxidizing agent (hydrogen peroxide; H2O2), availability of iodide and Tg, and correct spatial organization of these components (Fig. 1).
TPO is a heme-containing enzyme and is membrane-bound. It is active only in the presence of H2O2. There are two reactive sites on the enzyme, one for tyrosine and one for iodide. The iodide and tyrosine are thought to be oxidized in adjacent enzyme sites forming either a free radical of iodine or the iodinium ion (I+). The active iodine intermediate and the tyrosine are then covalently bound by TPO to form iodotyrosines (MIT, DIT). TPO also catalyses the peroxidative reaction leading to coupling of two iodotyrosines to form triiodothyronine (T3) and T4.[136]
The gene for TPO (human official gene symbol, TPO) is located on chromosome 2p24-p25 in humans. The gene contains 17 exons and 16 introns. The nucleotide sequence for the human (cDNA) TPO has been reported[137] and codes for a protein of 933 amino acids. This protein has five potential glycosylation sites and two disulfide bonds (creating a closed loop), and heme-binding sites.
Histidine 407 is the proximal heme-binding site in TPO and histidine 494 (and possibly 511, 520, 586) are the distal heme-binding sites. The proximal heme-binding site is coded by exon 8 of the (cDNA) TPO and the distal heme-binding sites are coded by exons 9 and 10 of the (cDNA) TPO.[137] Mutations affecting the TPO gene on these important sites will probably translate a biologically inactive TPO.
Alterations in thyroid hormones biosynthesis caused by abnormal function of the thyroid peroxidase system are summarized in Table 8 .
Iodide transported into the thyroid gland is rapidly oxidized by H2O2 and bound to tyrosine residues in Tg. Subsequently, iodinated tyrosine residues couple to form iodothyronine residues, mainly T4. Both iodination and coupling are catalyzed extracellularly by thyroid peroxidase at the apical border of the thyrocyte.
Concentration of iodide in the thyroid gland reaches a steady state between active influx, protein binding, and efflux, resulting in a relatively low intracellular iodine concentration under normal conditions. Iodide uptake is completely inhibited by anions of similar molecular size and charge, such as perchlorate or thiocyanate. On administration of sufficient concentration of these anions, iodide excess still present in the thyroid will be released into the circulation unless it is organified. The kinetics of iodide uptake and release can be traced by administration of radioiodine. After 131I administration, an analysis of the thyroidal iodide concentration in relation to that in serum and the degree of iodine bound to protein can be obtained. Partial iodide organification defects (PIOD) are characterized by release of less than 50% of the accumulated radioiodine. Total iodide organification defects (TIOD) are characterized by release of more than 90% of the radioiodine taken up by the gland within 1 hour after administration of sodium perchlorate. The radioiodide is usually given 2 hours before sodium perchlorate.
The common characteristic of these organification defects is a precipitous discharge of an isotopic iodide tracer from the thyroid after administration of perchlorate. At the clinical level, there is considerable clinical and biochemical heterogeneity. Most of the patients with TIOD (total iodine organification defect) have some degree of physical and mental retardation, large goiters, and hypothyroidism. In other cases there is compensation for the defect in organification of iodide by increasing the volume of thyroid tissue, forming large goiters that are able to maintain borderline euthyroidism, usually with elevated serum T3 levels and normal or low serum T4. The TSH response to TRH is exaggerated both in euthyroid and hypothyroid patients. In studies for which family pedigrees are available, the defect occurs in both genders, and in siblings. The unaffected parents are frequently consanguineous, thus giving a pattern of autosomal recessive inheritance.[138]
Criteria for the diagnosis of an organification defect, aside from the abnormal perchlorate discharge test, include the rapid uptake and release of radioiodine, low thyroidal iodine content, and elevated serum Tg levels that are raised to even higher serum concentrations after administration of bovine TSH. In vitro tests of TPO activity either by the triiodide method or the more physiologic test of incorporation of iodide into protein, reveals little or no TPO activity.[139] In a few patients, the TPO activity may be normal for organification of iodide, but is not effective in the coupling reaction within the Tg molecule. This is thought to be caused by an abnormally structured TPO.[138]
Molecular Genetics. Inactivating mutations in both TPO alleles have been found in patients with congenital hypothyroidism caused by to TIOD.[140] To date, approximately 20 different mutations have been described. Most mutations were found in exons 8, 9, and 10 containing the putative proximal and distal heme-binding histidine residues, the functional part of the TPO gene. The 1277-1278insGGCC in exon 8, leading to an early termination signal in exon 9, is the most frequently occurring mutation.[140] These authors presented the molecular characterization of 35 families with TIOD and detected structural defects in the TPO gene in one family confirming that most patients with TIOD have mutations in that gene.[5] In one patient with classic TIOD, a homozygous deletion in exon 14 was observed. The transmission pattern was anomalous and homozygosity appeared to be the result of partial maternal isodisomy of the short arm of chromosome 2 carrying the defective TPO gene.[141] The patient, born small for gestational age, appears healthy while being treated with T4. He has a normal phenotype, except for a unilateral preauricular skin tag. The single-base insertion in exon 14 resulted in a structurally altered TPO that could provide some degree of catalytic action in vivo. The molecular genetics studies explain the considerable clinical and biochemical heterogeneity that exists among patients with organification defect. In some patients, alternative splicing would generate a partially active form of the enzyme. In others, an early termination sign would prevent translation of the protein.[142,143,144,145,146] More recently Umeki et al.[147] described two novel mutations in the TPO gene, R665W and G771R in exons 11 and 13 respectively. The former was found in the patient's father (heterozygous) and the latter in her mother, also heterozygous. No TPO activity was detectable with cells transfected with mutated mRNAs. Moreover, the mutated TPO proteins showed abnormal cellular localization, exhibiting immunofluorescence only in the intracellular structure. Therefore the loss of apical membrane localization of the mutated TPO was the main cause for the iodide organification defect.
In five families with PIOD (defined as less than 50% iodide discharge after perchlorate), Nascimento et al.[148] found in one family compound heterozygosity in three affected patients, inherited, respectively, from both heterozygous parents. In the other four families only heterozygous TPO mutations and/or polymorphisms were detected. It is conceivable that the translated protein could be partially inactive and/or have an abnormal cellular localization.
Pendred's Syndrome. The association of goiter and congenital deafness was described by Vaughan Pendred in 1896,[2] in a family in which 2 of 5 children were deaf-mutes and had large goiters. Subsequent reports on families with Pendred's syndrome, many of them highly inbred, documented an autosomal recessive mode of inheritance. As first demonstrated by Morgans and Trotter in 1958,[149] the administration of perchlorate in these patients results in a partial discharge of radiolabeled iodide from the thyroid, indicating an impaired organification of this trace element to TG. Despite the presence of goiter and a mild organification defect, most patients with Pendred syndrome are euthyroid. Therefore, it is unlikely that a lack of thyroid hormones, which are essential for the development of the auditory system, are the cause of deafness in these individuals. The sensorineural deafness is typically but not always associated with a malformation of the inner ear, referred to as Mondini cochlea, in which the three-coiled structure of the cochlea is replaced by a single cavity in the apical region; hair-cells and ganglion cells are largely absent.[150,151] It was also documented that an enlarged vestibular aqueduct and endolymphatic duct is commonly associated with deafness in these patients.[150,151] Deafness is most commonly present at birth, but may only become apparent during childhood.
The incidence of Pendred's syndrome is thought to be as high as 7.5 to 10 in 100,000 individuals, because it has been estimated to account for about 10% of the cases with hereditary deafness. If these estimates are correct, Pendred's syndrome may be the most common form of syndromic deafness.
The Pendred's syndrome human gene official symbol SLC26A4 (solute carrier family 26, member 4; previous symbols DFNB4, PDS) encompassing 21 exons is located at chromosome 7q31[152,153] and codes for a protein of 780 amino acids called pendrin. Pendrin is located exclusively at the apical membrane of the follicular cell and presumably, its functional activity is to increase the influx of iodide into the follicular cell.[154] It is a member of a large family of anion transporters, predicted to contain 11 or 12 transmembrane domains. It was shown to transport iodide and chloride in a Na+-independent fashion.[155] Mutation analysis has been performed in a great number of families and consists of frameshift mutations, mutations leading to aberrant splicing processes and missense and nonsense mutations.[156,157,158,159,160,161] The L236P and the T416P proved to be recurrent mutations.
Thyroid NADPH Oxidase. The H2O2 generator comprises at least a membrane-bound flavoprotein that functions by transferring electrons from nicotinamide adenine dinucleotide phosphate (NAD(P)H) to molecular oxygen. DUOX1 (dual oxidase 1; human official gene symbol, DUOX1; aliases THOX1, NOXEF1) and DUOX2 (dual oxidase 2; human official gene symbol, DUOX2; aliases THOX2, LNOX2) are two novel large homologues of the NADPH oxidase flavoproteins Nox that are strongly expressed in the thyroid gland and at a lower level in some other tissues.[162,163,164] Accordingly, it was demonstrated that DUOX proteins in human thyroid tissues are located at the apical membrane of 40%-60% of normal thyrocytes.[162]
Their structure includes seven transmembrane-spanning domains, 3 NADPH and 1 FAD binding site and 2EF-hand motifs. DUOX2 is expressed at a higher level than DUOX1 in the thyroid, as determined by serial analysis of gene expression (SAGE). The DUOX1/DUOX2 genes are colocalized on 15q15.3 chromosome; DUOX2 is composed of 33 exons.
Moreno et al.[165] found mutations in the DUOX2 gene in one patient (patient 1) with TIOD and permanent CH and in 3 of 8 patients (patients 2, 3, and 4) with PIOD and transient CH. All mutations resulted in premature stop codons that delete the NADPH- and FAD-binding sites of the DUOX2 gene. In patient 1, exon 11 of DUOX2 had a homozygous substitution (1300T > C), which generates a premature termination signal (R434X). Patient 2 was heterozygous for the 2056T > C mutation in exon 16 of DUOX2, which also generates a premature stop codon instead of the incorporation of a glutamine (Q686X). Patient 3 was heterozygous for the 2101T > C nonsense mutation in exon 16 of DUOX2, which changes arginine 701 into a premature termination signal (R701X). Patient 4 was heterozygous for the 2895-2898delGTTC in exon 21 of DUOX2, which introduces a frame shift generating a termination signal in exon 22 (S965fsX994) (Fig. 5). However, these authors reported no functional studies to determine the implications of these mutations on PIOD. Figueiredo et al.[166] reported goiter and hypothyroidism in two siblings in family with no history of consanguinity caused by impaired H2O2-generating activity. Both patients had a positive perchlorate discharge tests (50% and 70%, respectively). Thyroid tissues obtained after surgery of the goiter had a normal thyroid peroxidase oxidation (1034-1064 U/g of protein) and a normal iodination of albumin (8-16 nmol/g protein). Thyroglobulin content was normal in both glands and could be normally iodinated by TPO in vitro. Thyroid NADPH oxidase activities were low in the particulate fraction, indicating that the H2O2 generation is the probable cause of the organification defect in these goiters (Fig. 6).

Functional domains and mutations of the thyroid oxidase 2 (THOX2) protein. Arrows indicate the places where mutant proteins are prematurely truncated. The frameshift induced by the S965fsX994 mutation codes for 29 aberrant amino acids before truncation. The relative position of calcium-binding (EF-hand), flavine adenine dinucleotide (FAD)-binding and nicotinamide adenine dinucleotide phosphate (NADPH)-binding motifs are indicated. (Reproduced with permission from Moreno JC, Bikker H, Kempers MJ, van Trotsenburg AS, Baas F, de Vijlder JJ, Vulsma T, Ris-Stalpers C 2002 Inactivating mutations in the gene for THOX2 and congenital hypothyroidism. N Engl J Med 347:95-102.)

Thyroid nicotinamide adenine dinucleotide phosphate (NADPH) oxidase activity in human thyroid from paranodular tissues from multinodular goiter patients without iodide organification defects (PN; n = 5) and from two siblings (DESM, DSM) with goiter and hypothyroidism because of an impaired iodide organification system. Note the decreased generation of H2O2 both in the particulate 3000 g (A) and 100,000 g (B) fractions (Reproduced with permission from Figueiredo MD, Cardoso LC, Ferreira AC, Campos DV, da Cruz Domingos M, Corbo R, Nasciutti LE, Vaisman M, Carvalho DP 2001 Goiter and hypothyroidism in two siblings due to impaired Ca++/NAD(P) H-dependent H2O2 generating activity. J Clin Endocrinol Metab 86:4843-4848.)
Defects in Thyroglobulin Synthesis. Thyroglobulin (human gene official symbol, TG) is the homodimeric (2 x 330,000 dd) glycosylated iodoprotein of sedimentation 19S that accumulates in the follicular lumen of thyroid follicles. Its abundance and degree of iodination vary greatly, depending on the activity of the gland.[167] TG is translated from an 8.4 kb mRNA encoded by a large (> 250,000 bp) transcription unit on the long arm of chromosome 8. The coding information is scattered among 42 exons. Following a 19-amino acid signal peptide, the polypeptide chain is composed of 2748 residues. Four hormonogenic peptides have been isolated from TG of various species, and two of them map to subterminal positions (residues 5 and 2746 in the human sequence). They correspond to sites involved preferentially in the synthesis of T4 and T3, respectively. Quantitative and qualitative abnormalities in (mRNA)TG, the intrathyroidal transport of TG, and its glycosylation, have been described in both animals and humans.[168,169,170,171,172,173,174,175,176,177,178,179,180,181,182,183,184,185] TG defects are transmitted in an autosomal recessive manner.
Animal Studies. Studies of a species of goiter with hereditary hypothyroidism demonstrated almost absent thyroidal Tg concentration. There was reduced (mRNA)Tg (< 10% of normal) and most of the small amount of (mRNA)Tg present was in the nucleus rather than in the endoplasmic reticulum. A strain of congenitally hypothyroid sheep has also been reported with a similar quantitative defect in (mRNA)Tg.[171] Absent 19S Tg and near absence of 12S Tg was reported in a strain of Afrikander cattle with goitrous hypothyroidism. In this bovine goiter an alternative splicing in (mRNA)Tg with absence of exon 9 is responsible for the defect.[170,172]
Congenital goiter in the (cog/cog) mouse is caused by mutations that in homozygous states cause primary hypothyroidism with goiter.[168] Several abnormalities in the Tg structure were reported in cog/cog Tg. Examination of thyrocyte ultrastructure from the mutant mice has strongly suggested that this illness represents a new member of the recently defined family of endoplasmic reticulum (ER) storage diseases, related to defective tissue-specific protein (Tg) export. Moreover it has been shown that the mutation in the coding sequences of Tg greatly inhibits protein dimerization and endoplasmic reticulum (ER) export.[173]
In contrast to the goitrous hypothyroidism present in the cog/cog mouse and certain human counterparts, the recessive dwarf rdw/rdw rat displays a nongoitrous form of primary CH.[5] The rdw/rdw rat also has a missense mutation in the TG gene replacing a highly conserved amino acid (G2320R). As in the cog/cog mouse, the mutated Tg shows a severe defect in export with retention inside the ER, but for not yet explained reasons, there is no goitrous enlargement of the thyroid. The findings in the rdw/rdw rat suggest that a subset of patients with dyshormonogenesis also may present with nongoitrous CH.[5]
Human Studies. In humans, quantitative abnormalities in TG synthesis are frequently found.[167] The radioiodine uptake is elevated and there is virtual absence of TG in both thyroid extracts and serum samples. A human recombinant TSH stimulation test fails to raise the serum TG concentration, and iodoalbumin is the chief iodoprotein detectable. Simultaneous serum measurement of protein-bound iodine and serum T4 demonstrates that protein-bound iodine (PBI) is greater than T4 iodine. Immunoelectrophoresis confirms the serum presence of non-19S TG iodinated proteins such as iodoalbumin or iodogammaglobulin, which may contain iodohistidine. TG-deficient patients also excrete in urine products probably originating from the breakdown of abnormal circulating iodoproteins that are being generated in the thyroid as low-molecular-weight iodinated material (LOMWIOM).[174] Microscopic examination of the thyroid gland has revealed a macrofollicular pattern with dilated follicles lined with high columnar cells and follicular lumen devoid of colloid, indicative of the absence of TG synthesis and long-term TSH stimulation. Many of the patients described with quantitative TG defects have normal serum levels of T3, which contribute to normal tissue metabolism. It is thus possible that small amounts of functionally active TG could be synthesized, iodinated, and immediately hydrolyzed, yielding mostly T3, as a result of the intense tissue stimulation by TSH.
Qualitative defects in TG have been described, when associated with structurally abnormal TG with a decrease in the ability of TG to be iodinated in vitro and a resistance to release of 125I when hydrolyzed in vitro. In other reported cases, Tg was found to be extremely labile and readily dissociated into subunits with reduced T3 and T4 content.[119] Further studies on the thyroid tissue of one affected hypothyroid goitrous sibling (JBM) from a family whose parents were first cousins reported to have six congenitally hypothyroid goitrous siblings were conducted by Grollman et al.[175] When the carbohydrates of normal and the defective TG were analyzed, the defective TG presented a sialic acid content remarkably low because of absence of sialyl transferase enzyme in the goiter tissue.[175] Severe hyposialylated TG is probably linked to a defect in iodotyrosine coupling and a possibly abnormal migration of TG into the follicular lumen.
Molecular Genetics. Several mutations in the TG gene responsible for cases of dyshormonogenesis have been described.[159,160,161,162,163] A hypothyroid woman with congenital goiter and marked impairment of TG synthesis has been studied by Ieiri et al.;[176] exon 4 was missing from the major TG transcript because of a cytosine to guanine transversion at position -3 in the splice acceptor site of intron 3.
Another TG mutation has been identified in a family with two affected siblings.[177] The thyroid (mRNA)TG was first reverse-transcribed, divided in overlapping segments, and the resulting cDNA products amplified by PCR. In the control normal thyroid tissue the amplification of nucleotide (nt) 4502 to nt 5184 showed a predominant fragment of 683 bp and a minor fragment of 512 bp. This latter fragment contained a 171-nt deletion. In the goitrous thyroid tissue, the predominant fragment was 512 bp. The sequence of the 683-bp fragment revealed that the responsible mutation is a C > T transition creating a stop codon at position 1510. This results in loss of a Taql restriction site. The point mutation is removed from a portion of the transcripts by the preferential accumulation in the goiter of a 171-nt-deleted (mRNA)TG. The reading frame is maintained and is potentially fully translatable into a TG polypeptide chain shorter by 57 residues. The presence of the deleted (mRNA)TG in normal thyroid tissue, albeit at a low level, strongly suggests that the deleted mRNA sequence corresponds to a complete exon that would contain the point mutation. It was concluded that the shorter, alternatively spliced (mRNA)TG predominates in the goitrous tissue and probably has a shorter life. The point mutation was removed by the preferential accumulation of the 171-nt-deleted (mRNA)TG indicating that there was a partial rescue providing an explanation for the relatively mild phenotype expression of the patients.
In another family[178] two siblings (HSN and AcSN) with congenital goitrous hypothyroidism had virtual absence of TG in the goitrous tissue. Sequencing of a fragment present in HSN thyroid tissue (mRNA)TG revealed that 138 nucleotides were missing between positions 5590-5727. This deletion does not affect the reading frame of the resulting mRNA and is potentially fully translatable in a TG polypeptide chain that is shorter by 46 residues. Extended molecular studies[183] of those two affected patients showed that the same 138-nt deletion was observed in RT-PCR studies performed in thyroid tissue from AcSN. In order to identify the intron/exon junction boundaries and to analyze the regions responsible for pre-mRNA processing corresponding to the 138-nt deletion, it was performed a screening of a human genomic library. The results showed that a deletion mapped between positions 5549 and 5586 of the (mRNA)TG and corresponded to exon 30. The positions of the exon limits differed by 3 nucleotides from the previously reported data because the intron/exon junctions in this region were not available at the time when the deletion was first described. The functional consequences of the deletion are related to structural changes in the protein molecule that either could modify the normal routing of the translation product through the membrane system of the cell or could impair the coupling reaction. Probably the mutant TG polypeptide might be functionally active in the production of thyroid hormone, as in the presence of normal iodide ingestion, AcSN was able to maintain normal T3 serum levels associated with relatively low serum T4 with normal somatic development without signs of brain damage.[183]
In a family with three affected individuals, the thyroid gland from one of the siblings was available for molecular studies.[179] TG messenger RNA was found at low levels while the (mRNA)TPO was found to be more abundant compared with control thyroid tissue. The low levels of (mRNA)TG was caused by a transcriptional defect due to the virtual absence of a thyroid-specific transcription factor (TITF1) expression as determined by Northern blot analysis, RT-PCR, and electrophoretic mobility shift assays.
In some families with affected siblings with defective TG synthesis no deletions or mutations have been found in (mRNA)TG obtained from the goitrous tissues.[180] Similarly, as has been demonstrated recently in the mutant mice (cog/cog), it is possible that point mutations in the coding sequences of TG would inhibit glycosylation and dimerization, causing defective ER export.[185]
In thyroid tissue from a 13-year-old patient suspected of a TG synthesis defect, the (mRNA)TG was studied. The complete coding region of 8307 bp was directly sequenced and revealed a homozygous point mutation: a C886T transition in exon 7 (Fig. 7). On translation this mutation would result in a stop codon at amino acid position 277, replacing the arginine residue. A (cDNA)TG construct containing the mutation was expressed in rabbit reticulocyte lysate resulting in a truncated protein of 30 kd, which maintained it glycosylation ability.[182] Two other siblings had a clinical presentation similar to the index patient, while their parents were unaffected. Additional restriction fragment length polymorphism analysis of the pedigree verified that the homozygous nonsense mutation cosegregated with the clinical phenotype. Clinically, hypothyroidism was not severe in the affected siblings because the truncated TG glycoprotein was still capable of thyroid hormonogenesis.

Impaired thyroglobulin (TG) synthesis in a consanguineous family with three affected siblings with goiter and hypothyroidism. The complete coding sequence of the TG gene cDNA was directly sequenced and revealed a homozygous point mutation (a C886T transition) in exon 7, resulting in a stopcodon at amino acid position 277. The truncated TG glycoprotein was still capable of thyroid hormonogenesis in the presence of excess iodine. (Modified from van der Graaf et al.[182]).
We have examined thyroid tissues from four patients with defective export of TG to the colloid.[185] Light microscopy demonstrated presence of intracellular TG despite its absence in thyroid follicle lumina, while electron microscopy indicated abnormal distention of the ER. It was confirmed that most intrathyroidal TG fails to reach the Golgi compartment, causing massive induction of specific ER molecular chaperones (Fig. 8). Thus, these kindreds suffer from a thyroid ER storage disease,[181] similarly to that found in cog/cog mice.[168]

Schematic representation of the consequences of defective thyroglobulin (TG) that leads to an endoplasmic reticulum (ER) storage disease causing congenital hypothyroid goiter. In affected patients the arrival of TG in the Golgi complex, and the subsequent secretion of TG to form tyroxine (T4) is drastically inhibited. Instead, TG accumulates in the ER, and accumulation of ER chaperones. The diminished synthesis of T4 triggers pituitary secretion of thyrotropin (TSH), further stimulating the thyroid gland and contributing to goiter growth
The screening for mutations in the (cDNA)TG from six patients with CH suspected of having a TG synthesis defect[186] did not show any major mutations in the two alternative splice transcripts identified, because all these transcripts were expressed in thyroid tissue of patients and controls. The authors concluded that the TG synthesis defect in these patients cannot be explained by major mutations in the coding region of the TG gene. This indicate the importance of a detailed, although still partial, knowledge of the structure of TG and its coding region of the extremely large wild-type human TG gene. As all reported TG mutations causing dyshormonogenesis in humans and animals are designated in the nucleotide and amino acid sequences, establishing the intron/exon organization of the human TG gene will help to understand the structure-function relationship in hereditary goiters with TG defects.[184]
Defects in Iodide Recycling. Deiodinase deficiency is another rare form of defective hormonogenesis in the thyroid gland. Removal of iodine from tyrosines is catalyzed by an iodothyronine dehalogenase, which is present in various mammalian tissues, including thyroid, kidney, and liver.[187] In the thyroid this enzyme has a special function, acting upon the iodotyrosines (monoiodotyrosine [MIT] and diiodotyrosine [DIT]) released during TG hydrolysis and liberating iodide, which can reenter the hormonogenesis pathway. Normally, deiodinating activity of this enzyme is so efficient that negligible amounts of iodotyrosines are secreted by the thyroid. Patients with congenital goitrous hypothyroidism caused by deficient dehalogenase activity are unable to deiodinate iodotyrosines, which are ultimately excreted in urine.[188] This urinary loss of iodotyrosines produces a chronic iodide deficiency, usually not compensated by iodide ingestion and goitrous hypothyroidism ensues. The reversal of hypothyroidism in several patients with this disorder by treatment with iodine supplements supports this view.
A number of cases, some in closely inbred families, were described by different authors.[187,188,189] The defect may be found to be total or partial (limited to the thyroid). In the total body defect, goitrous hypothyroidism and mental retardation are associated, the 24-hour 131I uptake is rapid; labeled DIT and MIT in increased amounts and low levels of T3 and T4 are found on serum chromatography. In the isolated intrathyroidal defect, subjects are goitrous but euthyroid, 24-hour radioiodine uptake and discharge are accelerated, but peripheral deiodination of labeled DIT and MIT is normal.
Diagnosis can only be made based on the presence of iodotyrosines and if tissue is available on enzyme activity, because the dehalogenase genes have not yet been cloned. The diagnostic criteria for the diiodotyrosine deiodinase defect are shown in Table 9 . In contrast to other inherited disorders of the thyroid system, this defect has not been explored at the molecular level.
Although the inheritance of the dehalogenation defect is considered to be autosomal recessive, some features of the disorder are expressed in heterozygous relatives, as goiter, a relatively high radioiodine uptake and increased urinary DIT excretion. The clinical expression strongly depends on the iodine content of the diet, which might explain why autosomal dominant inheritance has been suggested in some families.[190]
Thyroid Hormone Transport Protein Abnormalities. The three major transport proteins are TBG, transthyretin (TTR, formerly referred as thyroxine-binding prealbumin or TBPA) for the role it plays in the transport of retinol-binding protein),[191] and albumin.[10,11] TBG is functionally the most important T4-binding protein. It carries approximately 75% of the serum T4 and 70% of the serum T3. The hormones are associated with the transport proteins by noncovalent bonds and are in constant reversible equilibrium with free hormone (0.03% of total T4 and 0.3% of total T3). The free fraction of thyroid hormone is immediately available to tissues where it exerts its metabolic effects.
Abnormalities in the serum proteins that transport thyroid hormone do not alter the metabolic state or cause thyroid disease. However, they do produce alterations in thyroid hormone concentration in serum and when unrecognized may lead to inappropriate treatment.[192]
Clinically these defects usually manifest as either euthyroid hyperthyroxinemia or hypothyroxinemia and more rarely, hypertriiodothyroninemia.[193] Associated abnormalities such as thyrotoxicosis, goiter, and familial hyperlipidemia are usually coincidental.[11]
The existence of inherited defects of serum transport of thyroid hormone was first recognized in 1959 with the report of TBG excess by Beierwaltes and Robbins.[194]
TBG Defects. TBG is a 54-kd polypeptide that is synthesized in the liver and is encoded by a single gene copy. It is a member of the serine protease inhibitor (serpin) super-family of proteins. Proteolytic cleavage of TBG appears to be a mechanism for site-specific release of T4 independently of homeostatic control. TBG probably facilitates the transport of maternal T4 and iodide to the fetus, although this remains to be proven.[195]
Clinically, inherited abnormalities in TBG are classified according to the level of TBG in serum of the affected hemizygotes (XY males or XO females that express only the mutant allele) as: (1) complete deficiency, (2) partial deficiency, and (3) TBG excess.
Inheritance is X chromosome-linked[196] compatible with the location of the TBG gene on the long arm of the X-chromosome (Xq22.2)[197] and leads to the synthesis of an altered molecule devoid of hormone binding activity or secretion of an unstable molecule that is immediately degraded and removed. Some mutations (deletion in codon 352) may be specific for certain racial groups.[198]
Complete Deficiency of TBG. Complete TBG deficiency (TBG-CD) is defined as undetectable TBG in serum of affected hemizygous subjects or a value lesser than 0.03% the normal mean; the current limit of detection being 5 ng/dL by sensitive assays. In families with TBG-CD affected males have no detectable TBG, and heterozygous carrier females (mothers and daughters) usually express the defect partially by having approximately half the normal concentration of TBG.[6] TBG-CD is caused by either premature termination of translation or an amino acid substitution resulting in failure of secretion. It has been demonstrated that the mutant TBG may be retained within the rough ER favoring the intracellular degradation.[199] So far, at least 11 TBG-CD variants having this phenotype have been characterized at the molecular level.[200,201,202,203,204,205,206,207,208,209,210,211]
Partial Deficiency of TBG. Partial TBG deficiency (TBGPD) is the most common form of inherited TBG deficiency. For instance, approximately 50% of Australian Aborigines have an abnormal TBG (TBG-A),[212] and slow TBG (TBGS) is found in African and Pacific Island populations.[213] TBG-PD may be caused by several mechanisms. These include reduced secretion as a result of alterations in gene expression, abnormal intracellular processing, or accelerated rate of degradation caused by instability of the variant molecules.[11,198] At least, six different mutations, producing a variable degree of reduction of TBG concentration in serum, have been identified.[214,215,216,217,218,219,220,221,222,223,224] A unique family with TBG-PD has been recently described in which inheritance of the partial deficiency was autosomal dominant with transmission of the phenotype from father to son.[225] The serum concentration of TBG in affected males and females was approximately one half the normal mean value. The TBG had normal affinity for T4, normal isoelectric focusing and normal heat lability. No sequence changes were found in the entire coding area of the gene or in the promoter region. Although the mechanism of TBG-PD in this family is unknown an abnormality in one of the factors regulating TBG gene transcription might be considered as a possibility.
In families with partially TBG-deficient males, the mean TBG concentrations in heterozygous females is usually above half the normal, however, their identification by serum TBG measurement may be difficult because levels often overlap the normal range.
TBG Excess. Inherited TBG excess (TBG-E) is relatively rare cause of elevated levels of serum TBG. Serum TBG concentration in males with TBG-E is twofold to fourfold the normal mean, and in the corresponding carrier females is slightly higher than half that of the affected males. These observations indicate an equal contribution of cells expressing the normal and mutant TBG genes. For many years the molecular basis of TBG-E had been elusive and complete sequencing of the coding and promoter regions failed to show any defects.[226] However, in 1995, Mori et al.[227] found that gene amplification (duplication or triplication) was the cause of TBG-E in two Japanese families. Hemizygous affected males had approximately threefold and twofold the average normal serum TBG concentration, respectively. The presence of multiple TBG gene copies in tandem was confirmed by in situ hybridization of prometaphase and interphase chromosomes from an affected male. In another study, three Japanese families, one familial and two sporadic TBGE cases, were further analyzed by Mori et al.[228] Although amplification of TBG gene was not recognized in these three families by in situ hybridization of prometaphase chromosomes, the results confirmed that the affected X-chromosome presented three copies of the TBG gene. Similarly, a twofold increase in gene dosage was demonstrated in the affected sporadic cases males. This indicates that TBG-E is caused by gene duplication or triplication.
Transthyretin Defects. TTR is a tetrameric serum protein of 4 identical subunits of 127 amino acids each, synthesized mainly in the liver, the choroid plexuses of the brain and the retinal pigment epithelia of the eye.[229,230] Each monomer contains two beta sheets that interact to form a dimer. Its gene contains four exons. TTR formerly was called prealbumin because it migrates anodally to albumin on serum protein electrophoresis, but this name was misleading because TTR is not a precursor of albumin.
Serum TTR acts as a transport protein for T4 and also vitamin A;[231] in the latter case, this is accomplished by a formation of a 1:1 molar complex with retinol-binding protein. TTR binds virtually all of serum retinol-binding protein and approximately 15% of serum T4. TTR is the main T4 transport protein in cerebrospinal fluid (CSF) and might contribute to the transport of serum T4 across the blood-brain and blood-choroid plexus-CSF barriers.[232]
Screening of TTR gene on chromosome 18 (18q11.2-q12.1)[233] has uncovered mutations that produce variant TTR molecules with or without alterations in the binding affinity for iodothyronines.[234] Because TTR has a relatively lower affinity for T4 (approximately 100-fold lesser than that of TBG) it plays a minor role in thyroid hormone transport in blood.[235]
Accordingly, changes in the TTR concentration in serum and variant TTRs with reduced affinity for T4 have little effect on the concentration of serum T4.[234,236] Only variant TTRs with a substantially increased affinity for iodothyronines produce significant elevation in serum T4 and rT3 concentrations and account for 2% of subjects with euthyroid hyperthyroxinemia.[237,238,239,240,241,242,243,244,245,246,247]
A family with euthyroid hyperthyroxinemia with elevated total T4 concentration that was predominantly bound to TTR was first described in 1982 by Moses et al.[238] in eight subjects spanning three generations. The inheritance was autosomal-dominant, and affected members were clinically euthyroid with normal free T4 levels measured by equilibrium dialysis. The variant TTR has a single nucleotide substitution (A109T), which increases its affinity for T4, rT3 and triiodothyroacetic acid, and to a lesser extent, T3.[241,246] Crystallographic analysis of this variant TTR revealed an alteration in the size of the T4-binding pocket.[248] A more common defect found in subjects with prealbumin associated hyperthyroxinemia (PHA) is a point mutation in exon 4 of the TTR gene (T119M) that was first described in a single individual with normal serum total and free T4 levels in a North American kindred of Swedish ancestry. Harrison et al.[243] identified an apparently benign, electrophoretic variant of TTR. They identified a C > T point mutation in exon 4 (T119M). The variant was found incidentally in a girl with classic α-1-antitrypsin (AAT) deficiency and in her father during AAT phenotyping by an electrophoretic method. Five heterozygotes in three generations were studied. There was no evidence of amyloidosis in the family. The majority of subsequently identified heterozygous subjects harboring the TTR T119M had an increase in the fraction of T4 and rT3 associated with TTR, but only few had serum T4 levels above the upper limit of normal. Scrimshaw et al.[235] identified the same mutation (T119M) in four unrelated persons with euthyroid hyperthyroxinemia. The mutation created a new NcoI restriction endonuclease cleavage site that permitted its detection by a rapid and simple assay based on PCR. These authors[235] concluded that although the T119M mutation was associated with increased binding of T4, the hyperthyroxinemia in the patients who brought the variant to their attention appears to be transient, usually in association with nonthyroid illness. Alves et al.[244] found another family with this mutation during a screening program for TTR mutations in the Portuguese familial amyloidosis polyneuropathy (FAP) population. Cyanogen bromide peptide mapping and DNA RFLP analyses showed that the proband was a compound heterozygote for two TTR variants: H90A and T119M, inherited from the father and mother, respectively. Neither the compound heterozygote nor his parents had symptoms of FAP. They confirmed that TTR-binding of T4 was increased in association with the T119M mutation. Another TTR gene mutation has been more recently described by Refetoff et al.[247] They noted that of three TTR variants thus far identified with increased affinity for T4 (A109T, T119M, and G6S) only A109T has a high enough affinity to T4 to produce consistent hyperthyroxinemia in heterozygotes. Because the A109T mutation destroys a BsoFI site in exon 4 of TTR gene, use of this enzyme was suggested as a way to screen for A109T substitutions in subjects with euthyroid hyperthyroxinemia. These authors investigated a family with dominantly inherited euthyroid hyperthyroxinemia, for which 2 of 8 affected members had ablative thyroid treatment for presumed thyrotoxicosis and one was misdiagnosed as having resistance to thyroid hormones. All affected individuals had above normal serum rT3 levels, mean T4 levels 50% above those of their affected relatives and total T3 and TSH levels within the normal range. While the loss of the BsoFI site in TTR allele suggested the presence of an A119T substitution, sequencing of the TTR gene revealed a A109V mutation. Considering their (A109T, T119M, and A109V) association constant for T4-binding, it was concluded that the A109V variant has an increased affinity for T4 that approaches that of A109T, approximately 10-fold higher than that of wild-type TTR, and is sufficient to produce consistent hyperthyroxinemia in heterozygotes. A small portion of TTR mutations are apparently nonamyloidogenic. Among these are mutations responsible for hyperthyroxinemia.
TTR can undergo a conformational change and form amyloid fibrils in both acquired and hereditary forms of systemic amyloidosis. More than 80 mutations of the human TTR have been associated with autosomal dominant amyloidosis, usually presenting with peripheral and autonomic neuropathy and/or cardiomyopathy.[249,250] TTR mutations accelerate the process of TTR amyloid formation and are the most important risk factor for the development of clinically significant systemic familial amyloidosis. The age of symptom onset, pattern of organ involvement, and disease course vary but most mutations are associated with cardiac and/or nerve involvement. The gastrointestinal tract, vitreous and carpal ligament also are often affected. When the peripheral nerves are affected prominently, the disease is termed FAP. When the heart is involved heavily but the nerves are not, the disease is called familial amyloid cardiomyopathy (FAC). Regardless of which organ is involved most heavily, the general term is amyloidosis transthyretin type (ATTR). Most variants causing familial ATTR are rare, but a few are common in certain populations: V30M (Portugal, Japan, Sweden), V122I (African Americans and in some areas of West Africa), T60A (Irish and Irish Americans), L58H (United States, most commonly in mid-Atlantic region), and G6S (people of white European descent).[249,251,252] Noteworthy is that most nonamylogenic, as well as more frequent amylogenic, mutations such as V30M and V122I, occur in CpG dinucleotide hotspots.[253]
Human Serum Albumin Defects. Albumin is unglycosylated monomeric protein, molecular weight 65,000, which comprises approximately one half of the blood serum protein. Albumin functions primarily as carrier protein for steroids, fatty acids and thyroid hormones and play a role in stabilizing extracellular fluid volume. It is synthesized in the liver as preproalbumin, which has an N-terminal peptide that is removed before nascent protein is released from the rough ER. The product, proalbumin, is in turn cleaved in the Golgi vesicles to yield the secreted albumin. The human serum albumin (HSA) gene is 16,961 nucleotides long and is split into 15 exons by 14 introns.[254] HSA consists of a single polypeptide chain of 585 amino acid residues containing 17 disulfide bonds; the chain is folded into a heart-shaped molecule consisting of three homologous domains, each of which has two subdomains.[255] HSA is encoded on chromosome 4q11-q13[256] by a single gene. Mutations in HAS-encoding gene produce genetic variants (alloalbumins).[257] Most alloalbumins have a single amino acid substitution, resulting from a point mutation; few alloalbumins have a truncated and altered carboxyl terminus.[257] The phenotypes resulting from natural HSA mutants comprise the alloalbumins, the familial dysalbuminemic hyperthyroxinemia, bisalbuminemia, and analbuminemia. Alloalbuminemia is the term suggested for the variant albumins by Blumberg et al.[258] To date, more than 100 electrophoretic variants of HSA have been reported. Alloalbumins are benign and thus they are detected only by screening of blood proteins in studies of population genetics or during routine clinical electrophoresis. The mutations of the anomalous proteins vary in frequency, geographic distribution, and biologic stability.[259]
Familial Dysalbuminemic Hyperthyroxinemia. This condition, called familial dysalbuminemic hyperthyroxinemia (FDH) by Ruiz et al.[260]), is caused by increased affinity of a variant albumin for T4 and was first reported in 1979.[237,261] FDH phenotype is another form of dominantly inherited euthyroid hyperthyroxinemia. This condition is always associated with high serum total T4 levels and rarely with an increase in T3 and/or rT3 concentrations. The prevalence of FDH depends on the ethnic background of the population.[262,263,264] It is the most common cause of inherited increase of hyperthyroxinemia in the Caucasian population.[265,266] Recently described in a Japanese kindred[267] and in a Chinese individual,[268] no cases of FDH have been reported in subjects of African origin. The clinical significance of FDH is that standard laboratory methods for the estimation of free T4 or its direct measurement using T4 analogues give falsely elevated levels.[269] This has resulted in inappropriate surgical and drug therapy.[270,271,272]
The molecular basis of FDH was identified in 1994 by Sunthornthepvarakul et al.[265] and by Petersen et al.[266] A mutation in the ALB gene (R218H) produces an HAS with 10- to 15-fold higher affinity for T4 than the normal molecule and a 40-fold increase in the affinity for T3.[265,273] The identical mutation was found in 22 Caucasian families who were FDH tested.[265,266] Pohlenz et al.[274] reported a 5-month-old boy with FDH and congenital hypothyroidism who had a blood TSH level of 479 mU/L but normal T4 and elevated T3 levels. The patient and his euthyroid father and brother all carried the R218H mutation.
In 1997, Wada et al.[267] reported the first Asian family with FDH caused by a different mutation in the same amino acid (R218P). Although this variant HSA has 83-fold higher affinity for T4 than normal molecule, similar to the common R218H variant, the lesser increased in T3-binding affinity accounts for the modest increase of this hormone in serum of affected subjects.
A third mutation was reported in 1998 by Sunthornthepvarakul et al.[193] in the ALB gene (L66P) in a Thai family resulting in an HSA with 40-fold higher affinity for T3 but only 1.5-fold increase in the affinity to T4. As a consequence, patients have hypertriiodothyroninemia but not hyperthyroxinemia. The condition is also dominantly inherited and presents clinically as familial dysalbuminemic hypertriiodothyroninemia (FDH-T3) when T3 is measured by radioimmunoassay. More importantly, serum T3 concentrations are falsely low or even undetectable, when T3 is measured using an analog of T3 as a tracer rather than a radioisotope. FDH-T3 (L66P) is linked to the SacI (-) intragenic polymorphism and FDH A218H with SacI (+) polymorphism. This last association suggest a founder effect and is compatible with ethnic predilection of FDH.[265]
Bisalbuminemia. Bisalbuminemia is an asymptomatic variation of HSA. Heterozygotes have two species of albumin, a normal type and one that migrates abnormally rapidly or slowly on electrophoresis. T4 binding has been studied in subjects from unrelated families with a slow HSA variant. In two studies only the slow-moving HAS bound T4[275,276] and in another study, both bound T4.[277] Tarnoky and Lestas[276] described a new type of bisalbuminemia in two siblings and the son of one of them. The usual type was demonstrable by filter paper electrophoresis. The new type was demonstrable by electrophoresis on cellulose acetate at pH 8.6, but not on filter paper or starch gel. A phenocopy of hereditary bisalbuminemia, acquired bisalbuminemia, occurs with overdose of β-lactam antibiotics[278] and with pancreatic pseudocyst associated with pleural or ascitic effusion.[279] The anomalous albumin is anodal to the normal albumin in its electrophoretic mobility. In 1981, Vaysse et al.[280] described acquired trisalbuminemia in a patient with familial bisalbuminemia and pancreatic pseudocyst.
The differential binding of T4 to one of the components of bisalbumin may be because of enhanced binding to the variant component with charged amino acid sequence. Bisalbuminemia does not seem to be associated with gross alterations in thyroid hormone concentration in serum.
Analbuminemia. Analbuminemia is a rare autosomal recessive disorder manifested by the absence or severe reduction of circulating HSA in homozygous subjects, but heterozygous family members shown to carry the mutant gene have nearly normal levels.[281] Analbuminemia is readily detected by serum electrophoresis and is caused by a variety of mutations in the ALB gene. Previously, the mutation had been identified at the molecular level in only two human cases; in one case it resulted from an exon-splicing defect[282] and in the other case it was caused by a nucleotide insertion that caused a frameshift and premature stop codon.[281] In 1994, these authors[283] identified the mutations in three unrelated subjects from different countries. All three cases resulted from a single-nucleotide mutation that produced a stop codon, but the mutation occurred at different sites: (1) in an Italian male a 2368C > T transition in the genomic sequence of HSA; (2) a 4446C > T transition for an American female, and (3) a 7708G > A transition in a Canadian male. Campagnoli et al.[284] found in a male newborn of Iraqi extraction with analbuminemia a 118G > A substitution in the ALB gene. The mutation, involving the first base at intron 1, destroyed the GT dinucleotide consensus sequence found at the 5´ end of most intervening sequences and caused defective pre-mRNA splicing responsible for the analbuminemic trait. The child was homozygous; both parents were heterozygous. In 2002, Galliano et al.[285] described a new mutation associated with analbuminemia in four unrelated patients who had congenital analbuminemia: two of Amerindian origin and two of Turkish origin. In all four cases analbuminemia was caused by the same mutation (2430-2431delAT), the 91st and 92nd bases of exon 3. This novel defect, named Kayseri, produces a frameshift leading to a premature stop two codons downstream. The virtual absence of HSA is tolerable despite its multiple functions.
© 2003 Mary Ann Liebert, Inc.
Cite this: An Outline of Inherited Disorders of the Thyroid Hormone Generating System - Medscape - Aug 01, 2003.