
Abstract
Use of microbes to produce liquid transportation fuels is not yet economically viable. A key point to reduce production costs is the design a cell factory that combines the continuous production of drop-in fuel molecules with the ability to recover products from the cell culture at low cost. Medium-chain hydrocarbons seem ideal targets because they can be produced from abundant fatty acids and, due to their volatility, can be easily collected in gas phase. However, pathways used to produce hydrocarbons from fatty acids require two steps, low efficient enzymes and/or complex electron donors. Recently, a new hydrocarbon-forming route involving a single enzyme called fatty acid photodecarboxylase (FAP) was discovered in microalgae. Here, we show that in illuminated E. coli cultures coexpression of FAP and a medium-chain fatty acid thioesterase results in continuous release of volatile hydrocarbons. Maximum hydrocarbon productivity was reached under low/medium light while higher irradiance resulted in decreased amounts of FAP. It was also found that the production rate of hydrocarbons was constant for at least 5 days and that 30% of total hydrocarbons could be collected in the gas phase of the culture. This work thus demonstrates that the photochemistry of the FAP can be harnessed to design a simple cell factory that continuously produces hydrocarbons easy to recover and in pure form.
Similar content being viewed by others

Introduction
Increasing costs of petroleum extraction and environmental concerns over petroleum production and use have emphasized the need to develop renewable transportation fuels using energy-rich biomolecules such as alcohols, fatty acids and hydrocarbons1,2. Production of high titers of such carbon-based molecules has been achieved using heterotrophic or photosynthetic microbes but several impediments to commercialization of advanced biofuels remain3,4,5,6,7,8. For instance, some biomolecules like butanol can only be used as blending agents to conventional diesel fuel9 and others such as fatty alcohols or free fatty acids would require extraction from the fermentation broths. Still others, like fatty acid glycerol esters (oils), require expensive and energy-costly steps consisting in biomass harvest, conversion to biocrude and refinement, or oil extraction and chemical transesterification10.
In order to reduce drastically the costs of an industrial process, direct synthesis of fuel-like molecules by microbes combined with their release and capture outside of the cells is highly desired. One of the most promising types of fuel-like molecules appears to be hydrocarbons (HCs), especially n-alkanes, which are major components of current fossil fuels. Indeed, these compounds and their unsaturated analogs (n-alkenes) can be derived from abundant cell components (fatty acids) and, unlike oils, they are volatile or semi-volatile in the C5-C16 chain length range, which may be key to promote their release outside cells11,12,13. Formation of C9-C17 alka(e)nes by genetically modified microorganisms expressing HC-forming enzymes from a variety of organisms has been reported in bacterial and yeast cells14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29. Despite these advances, industrial prospects of microbial HC production seem limited by the competition of various host reductases for fatty intermediates of many HC-forming enzymes14,17,21,30 and the low turnover and complex cofactor requirement of the HC biosynthesis enzymes discovered so far15,26,31,32,33,34. Besides, recovery and quantification of liquid HCs in the gas phase of the cultures was only addressed in a single study but without estimation of productivity (mg L−1 h−1)17.
Recently, a novel HC-forming pathway has been discovered in the microalga Chlorella variabilis NC64A35. The HC-forming enzyme has been identified and shown to be a flavin-bearing protein that converts free fatty acids (FFA) into alkanes or alkenes36. This enzyme has been named fatty acid photodecarboxylase (FAP, EC 4.1.1.106) because it catalyzes the decarboxylation of free fatty acids (FFAs) using light (blue photons) to drive the reaction. It is thus a photoenzyme, a rare type of catalyst37. In vitro, the Chlorella FAP is known to accept a broad range of FFAs from C2 to C2236,38. FAP is likely to be of great interest for production of fuel in microorganisms because the one step conversion of fatty acids into HCs does not involve any intermediates such as aldehydes. In addition, FAP will not compete with other enzymes for chemical energy or cofactors as the reaction is electron-neutral and uses only light as energy source. A recent comparison of FAP with the widely used cyanobacterial aldehyde deformylating oxygenase (ADO) has shown that heterologous expression of the former in cyanobacteria increased HC production by 19-fold compared to the latter39. However, the potential of FAP to produce volatile HCs in the gas phase of cell cultures has not yet been investigated. Here our purpose is to address three questions: (i) What is the production rate of HCs in the gas phase of cell cultures expressing a HC-forming enzyme (compared to the production in cells)? (ii) Are HCs produced only transiently or is it possible to obtain a sustained production? (iii) What is the influence of light intensity on HC production in a FAP-expressing bacterial factory?
Results
Light dependency of FAP-based hydrocarbon production in E. coli
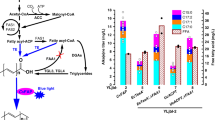
Because fatty acid photodecarboxylase (FAP) is a photoenzyme and its activity increases with white light intensity36, light may be a limiting factor of HC production in E. coli. HC production was analysed in batch cultures of an E. coli strain expressing FAP exposed to various white light intensities in order to determine saturating light. While this strain did not produce measurable HC amounts in the dark, growing cells under low to medium light intensity (50 to 150 µmol photons m−2 s−1) triggered maximal HC production (Fig. 1A). Increasing light intensity up to 500 µmol photons m−2 s−1 resulted in a decreased production of HCs per liter of culture medium, which was even more drastic at 1000 and 1500 µmol photons m−2 s−1. Clearly, this drop in HC yield was correlated with a strong decrease in the growth of the bacterial culture (Fig. 1B). Measurement of the dioxygen content in the culture medium using either a Clark electrode or a Membrane Inlet Mass Spectrometry (MIMS) showed that a quick decrease in dioxygen concentration occurred when the TB medium was exposed to blue light (Supplemental Fig. 1). Depletion of dioxygen in the medium culture was thus likely to be responsible for reduction of E. coli growth under high light. Interestingly, HC production at 48 hours post-induction dropped by 50% although cell growth dropped only by 20%. This indicated that cell growth was not the only cause of the drop in HC production under high light. We therefore checked the level of FAP expression by immunodetection in the soluble proteins extracted from E. coli cells exposed to different light intensities. FAP amounts present in the E. coli FAP-expressing strain decreased with increasing light intensity whereas the flavodiiron protein (FLV), another flavoprotein used as a control, remained constant (Fig. 1C,D and Supplemental Fig. 2). This result shows that under high light the reduction in growth of E. coli cultures does not affect protein expression under the T7 promotor and suggests that he FAP is likely photo-damaged when exposed to an excess of light. Since the FAP catalytic cycle involves a radical form of FAD36, a lack of substrate and an excess of light may indeed lead to accumulation of free radicals and inactivation of the FAP. Taken together, these experiments therefore show that in an E. coli FAP-based HC production system, maximum productivity is reached at relatively low light irradiance.

Influence of light intensity on HC production, bacterial growth and level of soluble FAP in a FAP-expressing E. coli strain. Strains were grown in batch culture under various light intensities. Data represent the mean ± SD of three independent experiments using three different bacterial cultures. (A) Relative HC production measured in cells 48 hours after induction. Production per culture volume was normalized by maximum obtained. (B) Growth of the culture measured by optical density (OD) at 600 nm, 24 hours or 48 hours post-induction. (C) SDS-PAGE of soluble proteins extracted from an E. coli strain expressing the FAP or a control strain expressing another flavoprotein (flavodiiron protein, FLV). 0, 500 and 1500 correspond to light intensities in µmol photons m2 s−1. Equal protein amounts were loaded in each lane. Bands of FAP and FLV are indicated by arrows. (D) Western blot of the SDS-PAGE revealed with anti-FAP or anti-FLV antibodies. Full-length gel and blots are included as Supplemental Fig. 2.
Set-up of a system to grow bacteria expressing FAP under light and collect hydrocarbons
With a view to design the simplest cell factory producing a drop-in fuel that could be easily recovered, we assessed the potential of FAP to produce medium-chain n-alkanes and n-alkenes (C9-C13) in the gas phase of the cultures. A strain coexpressing the FAP and a fatty acid thioesterase from the plant Umbellularia californica (Tes) was constructed (Fig. 2). The thioesterase Tes interrupts fatty acid elongation by hydrolyzing specifically medium-chain fatty acids esterified to acyl carrier protein and releasing medium-chain free fatty acids40. In order to investigate production of HCs by the FAP on a larger scale, E. coli cells were cultivated in transparent glass bottles bubbled with air and illuminated with medium-light intensity white LED panels (Fig. 3). In larger scale cultures, each cell is subjected to a fluctuating light due to constant agitation and the shading effect. Therefore, incident light cannot be easily correlated with actual light received by cells. We thus set up the incident light, so that the light intensity measured in the cell culture was within the optimal range determined on the smaller scale cultures (50–150 μmol photons m−2 s−1). An incident light intensity of 500 µmol photons m−2 s−1 was chosen because it was found to result in an illumination of around 100 µmol photons m−2 s−1 in the bulk of the cell culture. To quantify the fraction of alka(e)nes present in the gas phase of the culture, a HC-trapping device consisting of a low-cost matrix used in industry (activated charcoal) was placed at the gas outlet of the cultivation device. This system thus allowed to investigate HC production in cells as well as the gas phase of E. coli strains expressing the FAP.

Metabolic reactions used for photoproduction of hydrocarbons by E. coli cells. Two strains were cultivated. One was transformed with a gene coding a fatty acid photodecarboxylase (FAP) from Chlorella. The other one was transformed with genes coding FAP and a medium-chain specific thioesterase (Tes) from Umbellularia. Both genes are expressed under inducible promotors. FAP requires light as a cofactor. ACP: acyl carrier protein.

Scheme of the cultivation system used for photoproduction of hydrocarbons by E. coli cells. Air input was set at 15 L h−1 because this flow rate allowed proper oxygenation of the culture while minimizing foaming (too much foam resulted in partial wetting of the hydrocarbon trap). In controls experiments, a second trap connected on the outlet of the first one allowed to show that potential losses represented less than 1% of total hydrocarbons found in the first trap. Light intensity was about 100 µmol photon m2 s−1 in the culture, corresponding to 500 µmol photon m2 s−1 incident light.
Combination of FAP and Tes allows continuous production of volatile hydrocarbons
The FAP of C. variabilis was first expressed alone under a lactose-inducible promotor. Following induction of FAP expression, HC production reached a pseudo-plateau within two days, with 22 mg L−1 of HCs in the cell fraction and 26 mg L−1 in total (Fig. 4). HCs produced were mainly composed of C15:0 alkane and C17:1 alkene (20% and 75% respectively of the total HC production), with shorter chain HCs present in smaller amounts. Not surprisingly, the gas phase was enriched in shorter HCs, 45% of the total being C11 and C13 HCs (Fig. 5). As the culture aged, the relative content of short-chain HCs in the cells decreased, which was consistent with the idea that shorter HCs diffused to the gas phase.

Hydrocarbon production in cultures of E. coli expressing FAP or FAP and thioesterase. Time course of hydrocarbon production. Cells were grown in batch culture under constant light illumination. Chlorella variabilis FAP (FAP) and Umbellularia californica thioesterase (Tes) were expressed in E. coli. Hydrocarbons were quantified in the harvested cells and in the gas phase of the culture using the hydrocarbon trap (Fig. 2). Total refers to cells plus gas phase. Data represent the mean ± SD of three independent experiments using three different bacterial cultures.

Profile of hydrocarbons produced in E. coli cultures. Production of E. coli strains expressing FAP (FAP) or co-expressing FAP and thioesterase (Tes FAP) (A) Profile of hydrocarbons produced in E. coli cultures 120 h after induction. (B) Relative hydrocarbons composition of cell content in function of time (hours). Data represent the mean ± SD of three independent experiments using three different bacterial cultures.
In order to boost the production of medium-chain HCs, the FAP was then coexpressed with the thioesterase Tes. The gene encoding Tes was driven by an arabinose-inducible promoter to avoid overproduction of free fatty acids. When expressed alone in E. coli, Tes induced a 2-fold increase in the total fatty acid content of cells (Fig. 6A), which was mainly due to an increase in C12:0 and C14:1 but also C18:1 fatty acids (Fig. 6B). This showed that Tes was effective at enriching cells in medium-chain fatty acids and boosting fatty acid synthesis in general. Coexpression of Tes with FAP resulted in a striking 4-fold increase in HC production compared to the strain expressing only FAP, reaching a titer of 94 mg L−1 at day 5 (Fig. 4). Production of HCs in gas phase also significantly increased to 29 mg L−1 of HC, representing 30% of total HC production. HC profile showed an increase in medium-chain HC species with 15% of the production in the cell comprised of C11-C13 HCs compared to less than 2% without Tes (Fig. 5B). In the gas phase, the percentage of C11:0 alkane reached more than 30% of total HCs and C11 to C13 HCs represented more than 60% of the total HC production. Most importantly, the production rate from day one to day two reached 0.88 mg L−1 h−1 and remained constant at 0.91 mg L−1 h−1 from day two to day five. In comparison, without the thioesterase, production reached 0.55 mg L−1 h−1 until day two but it dropped down to 0.13 mg L−1 h−1 between day two and five. Therefore, HC production in a strain expressing only the FAP stopped as the bacteria reached stationary phase. By contrast, in the presence of the heterologous expressed thioesterase, fatty acid synthesis was kept active even in stationary phase, presumably to keep constant the pool of acyl-ACPs, which was continuously depleted by conversion to FFAs and HCs.

Fatty acid content in E. coli strain expressing Umbellularia californica thioesterase. (A) Total fatty acid content. (B) Content in individual fatty acids. After whole cell transmethylation, cellular fatty acids were quantified as fatty acid methyl esters (FAME) and expressed as mg FAME per liter of culture. A strain expressing a pBAD empty vector (EV) and a strain expressing pBAD-Thioesterase (Tes) were analyzed. Data represent the mean ± SD of three independent experiments using three different bacterial cultures.
Discussion
Here we describe a simple FAP-based cell factory that continuously releases volatile hydrocarbons and we quantify the production rates in the cells and in the gas phase of the cultures. We also show that low to medium light intensities (50–150 µmol photons m−2 s−1) allow to reach maximal HC productivity and that higher irradiances result in decreased FAP amounts.
Despite little metabolic engineering, HC productivity measured for the FAP-Tes strain (0.91 mg L1 h−1) was in the same range as the best ones that could be estimated for alkanes/alkenes production from previous studies, i.e. around 1–2 mg L−1 h−1 14,20. Additional genetic modifications such as arrest of fatty acid β-oxidation or increase of FFA pool17,41 may further boost HC production. But the key point is made here that the thioesterase from Umbellularia californica not only enriches FAP products in medium-chain HCs that are volatile, but is also key to achieve a continuous production of HCs while cells are not dividing anymore. Besides, the absence of aldehyde intermediates allows to recover HCs in pure form and not as a blend with fatty alcohols and fatty aldehydes14,17.
This work therefore shows that coexpression in E. coli of the newly discovered photoenzyme FAP and a medium-chain thioesterase results in a light-driven synthesis of medium-chain alkanes and alkenes, which can be easily recovered from the gas phase of bacterial culture in substantial amounts and highly pure form. Most importantly, evidence is provided that the production of volatile HCs by bacterial cells can be maintained at a high level over a period of several days. Therefore, this study demonstrates that the combination of FAP and thioesterase is a simple but powerful tool to turn E. coli into a cell factory that continuously releases volatile HCs. It should thus provide useful information for a bio-based production of fuel-range HCs on a larger scale.
Methods
Strains and cultivation conditions
E. coli BL21 pRIL strain was used throughout this study. Strains obtained by transformation of BL21 cells are listed in Supplemental Table 1. Pre-cultures were incubated at 37 °C overnight in Luria-Bertani (LB) broth medium with the following antibiotics, kanamycin 50 µg mL−1, chloramphenicol 34 µg mL−1, ampicillin 100 µg mL−1, depending on strain used. Cultures were performed in 24-deepwell plates for light intensity analysis, in 100 mL erlens for FAP expression analysis and in 500 mL bottles with a bubbling system for volatile HC analysis (containing 4, 30 or 300 mL of culture medium respectively). Light was provided by white LED panels. Terrific Broth (TB) medium (12 g L−1 tryptome, 24 g L−1 yeast extract, 12.5 g −1 K2HPO4, 2.3 g L−1 KH2PO4) with addition of required amounts of antibiotics was inoculated using a pre-culture (dilution 1:50) and grown at 37 °C at 180 rpm. When optical density (OD) at 600 nm reached around 0.8, cultures were induced by addition of isopropyl β-D-1-thiogalactopyranoside (IPTG) at a final concentration 0.5 mM or/and by addition of arabinose at a final concentration of 0.2% (w/v). Upon induction, temperature was lowered to 22 °C and culture were illuminated with white LED panels set at various light intensities (50 to 1500 µmol photon m2 s−1 incident light). For cultures in bottles, a stirring bar and an air bubbling system allowed mixing as well as gas exchange of the culture, air flow was run continuously at 15 L h−1 and the air outlet was connected to cartridges containing activated charcoal to trap HCs on exit.
Gene synthesis and construction of plasmids
Synthetic genes codon-optimized for E. coli were designed for Chlorella fatty acid photodecarboxylase (FAP) (Sorigué et al. 2017) and for the thioesterase (Tes) from the plant Umbellularia californica (Voelker and Davis 1994). Sequences of FAP and Tes corresponded to the proteins without their predicted chloroplast transit peptide (Supplemental Fig. 3). The plasmid expressing FAP was constructed from pLIC07 by Golden Gate cloning using BsaI restriction enzyme (Supplemental Fig. 4). pLIC07 is a vector derived from pET28 by insertion of SacB gene with restriction site for ligation independent cloning (LIC) (Supplemental Fig. 3). For co-expression, the plasmid expressing Tes was constructed from pBAD/Myc-his B by In-Fusion®. Primers used for plasmid construction are listed in Supplemental Table 2.
Extraction of hydrocarbons and fatty acids
For quantification of HCs contained inside the cells, one mL of culture was pelleted by centrifugation in glass tubes. Transmethylation was conducted by adding 2 mL of methanol containing 5% (v/v) sulfuric acid to the cell pellet. Internal standards (10 µg of hexadecane and 20 µg of triheptadecanoylglycerol) were added for quantification. Reaction was carried out for 90 min at 85 °C in sealed glass tubes. After cooling down, one mL of 0.9% (w/v) NaCl and 500 µL of hexane were added to the samples to allow phase separation and recovery of fatty acid methyl esters (FAMEs) and HCs in the hexane phase. Samples were mixed and then centrifuged to allow phase separation. One µL of the hexane phase was injected in the GC-MS/FID. For quantification of HCs trapped in the air outlet (i.e. the gaseous phase of the culture), the activated charcoal was incubated at room temperature in 2 mL of hexane containing10 µg of hexadecane as an internal standard. Samples were centrifuged for 10 min to pellet charcoal. One µL of the hexane phase was injected in the GC-MS/FID.
GC-MS/FID analyses
Analyses were carried out on an Agilent 7890A gas chromatographer coupled to an Agilent 5975 C mass spectrometer (simple quadrupole). A Zebron 7HG-G007-11 (Phenomenex) polar capillary column (length 30 m, internal diameter 0.25 mm, and film thickness 0.25 mm) was used. Dihydrogen carrier gas was at 1 mL min−1. For samples obtained from whole cell transmethylations, oven temperature was programmed with an initial 2-min hold time at 35 °C, a first ramp from 35 to 170 °C at 15 °C min−1, followed by 1-min hold time at 170 °C then a second ramp from 170 to 240 °C at 5 °C min−1 and a final 2-min hold time at 240 °C. For samples recovered from activated charcoal, oven temperature was programmed with an initial 2-min hold time at 35 °C, a first ramp from 35 to 180 °C at 15 °C min−1 followed by 2-min hold time at 180 °C. Samples were injected in splitless mode (1 min) at 250 °C. The MS was run in full scan over 40 to 350 amu (electron impact ionization at 70 eV), and peaks of FAMEs and HCs were quantified based on the FID signal using the internal standards C17:0 fatty acid and hexadecane respectively.
Protein analysis
Total proteins were extracted from cell pellets using a Bugbuster® protein extraction reagent. After 30 min extraction at room temperature, the supernatant was denaturated by incubation at 70 °C for 20 min in NuPAGE LDS sample buffer with 1 mM dithiothreitol. Total protein extract were separated on a 15% (w/v) acrylamide Bis-Tris gel using a Tris Glycine SDS buffer. After staining with ProSieveTM EX Safe Stain, quantification of total protein content in gel was done using an infrared imaging scaner measuring 700 nm fluorescence. For immunoblot analysis, proteins were loaded on a constant protein basis, separated as described before and then transferred with a semi-dry technic onto a BioTrace NT nitrocellulose membrane (Sigma-Aldrich). Membrane was blocked overnight at 4 °C by incubation with 5% (w/v) dried milk in Tris Buffer Saline containing 0.1% (w/v) Tween 20. Membrane was then incubated at room temperature for 2 h with specific primary polyclonal antibodies from rabbit (dilution 1:2000) and then for 1 h with a secondary anti-rabbit antibody coupled to horseradish peroxidase (HRP). Immobilon TM Western Chemiluminescent HRP substrate (EMD Millipore) was used for detection and images were recorded using a G:BOX Chemi XL (Syngene).
Dioxygen measurement
O2 exchanges were measured using a Clark-type electrode (S1 dioxygen electrode, Hansatech), or a Membrane inlet mass spectrometry system42. Blue (460 nm) or red (620 nm) light was applied on top of the cuvette using a Dual-PAM 100 module (Walz). The Clark electrode was set so that ambient O2 concentration in water (236 µmol l−1) gives a signal of 1 V, and absence of O2 (reached using reduced dithionite) gives a signal of 0 V. O2 concentration was recorded continuously upon red or blue illumination in both systems for a few minutes.
References
Marella, E. R., Holkenbrink, C., Siewers, V. & Borodina, I. Engineering microbial fatty acid metabolism for biofuels and biochemicals. Curr. Opin. Biotechnol. 50, 39–46 (2018).
Beller, H. R., Lee, T. S. & Katz, L. Natural products as biofuels and bio-based chemicals: fatty acids and isoprenoids. Nat. Prod. Rep. 32, 1508–26 (2015).
Wijffels, R. H. & Barbosa, M. J. An outlook on microalgal biofuels. Science 329, 796–9 (2010).
Wijffels, R. H., Kruse, O. & Hellingwerf, K. J. Potential of industrial biotechnology with cyanobacteria and eukaryotic microalgae. Curr. Opin. Biotechnol. 24, 405–13 (2013).
Desai, S. H. & Atsumi, S. Photosynthetic approaches to chemical biotechnology. Curr. Opin. Biotechnol. 24, 1031–6 (2013).
Runguphan, W. & Keasling, J. D. Metabolic engineering of Saccharomyces cerevisiae for production of fatty acid-derived biofuels and chemicals. Metab. Eng. 21, 103–113 (2014).
Pattanaik, B. & Lindberg, P. Terpenoids and their biosynthesis in cyanobacteria. Life 5, 269–93 (2015).
Meadows, C. W., Kang, A. & Lee, T. S. Metabolic engineering for advanced biofuels: production and recent advances toward commercialization. Biotechnol. J. 13, 1600433, https://doi.org/10.1002/biot.201600433 (2018).
Harvey, B. G. & Meylemans, H. A. The role of butanol in the development of sustainable fuel technologies. J. Chem. Technol. Biotechnol. 86, 2–9 (2011).
Delrue, F. et al. Comparison of various microalgae liquid biofuel production pathways based on energetic, economic and environmental criteria. Bioresource Technol. 136, 205–212 (2013).
Jetter, R. & Kunst, L. Plant surface lipid biosynthetic pathways and their utility for metabolic engineering of waxes and hydrocarbon biofuels. Plant J. 54, 670–83 (2008).
Wang, W. & Lu, X. Microbial synthesis of alka(e)nes. Front. Bioeng. Biotechnol. 1, 10, https://doi.org/10.3389/fbioe.2013.00010 (2013).
Herman, N. A. & Zhang, W. Enzymes for fatty acid-based hydrocarbon biosynthesis. Curr. Opin. Chem. Biol. 35, 22–28 (2016).
Schirmer, A., Rude, M., Li, X., Popova, E. & del Cardayre, S. B. Microbial biosynthesis of alkanes. Science 329, 559–62 (2010).
Rude, M. et al. Terminal olefin (1-alkene) biosynthesis by a novel P450 fatty acid decarboxylase from Jeotgalicoccus species. Appl. Environ. Microbiol. 77, 1718–27 (2011).
Howard, T. P. et al. Synthesis of customized petroleum-replica fuel molecules by targeted modification of free fatty acid pools in Escherichia coli. Proc. Natl. Acad. Sci. USA 110, 7636–41 (2013).
Choi, Y. J. & Lee, S. Y. Microbial production of short-chain alkanes. Nature 502, 571–4 (2013).
Kaiser, B. K. et al. Fatty aldehydes in cyanobacteria are a metabolically flexible precursor for a diversity of biofuel products. PLoS One 8, e58307, https://doi.org/10.1371/journal.pone.0058307 (2013).
Rui, Z. et al. Microbial biosynthesis of medium-chain 1-alkenes by a nonheme iron oxidase. Proc. Natl. Acad. Sci. USA 111, 18237–42 (2014).
Liu, Y. et al. Hydrogen peroxide-independent production of α-alkenes by OleTJE P450 fatty acid decarboxylase. Biotechnol. Biofuels 7, 28, https://doi.org/10.1186/1754-6834-7-28 (2014).
Coursolle, D., Lian, J., Shanklin, J. & Zhao, H. Production of long chain alcohols and alkanes upon coexpression of an acyl-ACP reductase and aldehyde-deformylating oxygenase with a bacterial type-I fatty acid synthase in E. coli. Mol. BioSyst. 11, 2464–2472 (2015).
Song, X., Yu, H. & Zhu, K. Improving alkane synthesis in Escherichia coli via metabolic engineering. Appl. Genet. Mol. Biotechnol. 100, 757–767 (2016).
Cao, Y. X. et al. Heterologous biosynthesis and manipulation of alkanes in Escherichia coli. Metab. Eng. 38, 19–28 (2016).
Patrikainen, P., Carbonell, V., Thiel, K., Aro, E. M. & Kallio, P. Comparison of orthologous cyanobacterial aldehyde deformylating oxygenases in the production of volatile C3-C7 alkanes in engineered E. coli. Metab. Eng. Commun. 5, 9–18 (2017).
Lehtinen, T., Virtanen, H., Santala, S. & Santala, V. Production of alkanes from CO2 by engineered bacteria. Biotechnol. Biofuels 11, 228 (2018).
Bernard, A. et al. Reconstitution of plant alkane biosynthesis in yeast demonstrates that Arabidopsis ECERIFERUM1 and ECERIFERUM3 are core components of a very-long- chain alkane synthesis complex. Plant Cell 24, 3106–18 (2012).
Chen, B., Lee, D. Y. & Chang, M. W. Combinatorial metabolic engineering of Saccharomyces cerevisiae for terminal alkene production. Metab. Eng. 31, 53–61 (2015).
Zhou, Y. J. et al. Production of fatty acid-derived oleochemicals and biofuels by synthetic yeast cell factories. Nature Comm. 7, 11709, https://doi.org/10.1038/ncomms11709 (2016).
Kang, M. K., Zhou, Y. J., Buijs, N. A. & Nielsen, J. Functional screening of aldehyde decarbonylases for long-chain alkane production by Saccharomyces cerevisiae. Microb. Cell Fact. 16, 74, https://doi.org/10.1186/s12934-017-0683-z (2017).
Rodriguez, G. M. & Atsumi, S. Toward aldehyde and alkane production by removing aldehyde reductase activity in Escherichia coli. Metab. Eng. 25, 227–237 (2014).
Andre, C., Kim, S. W., Yu, X.-H. & Shanklin, J. Fusing catalase to an alkane-producing enzyme maintains enzymatic activity by converting the inhibitory byproduct H2O2 to the cosubstrate O2. Proc. Natl. Acad. Sci. USA 110, 3191–3196 (2013).
Khara, B. et al. Production of propane and other short-chain alkanes by structure-based engineering of ligand specificity in aldehyde-deformylating oxygenase. Chembiochem. 14, 1204–8 (2013).
Zhang, J., Lu, X. & Li, J. J. Conversion of fatty aldehydes into alk (a/e)nes by in vitro reconstituted cyanobacterial aldehyde-deformylating oxygenase with the cognate electron transfer system. Biotechnol. Biofuels 6, 86, https://doi.org/10.1186/1754-6834-6-86 (2013).
Hayashi, Y., Yasugi, F. & Arai, M. Role of cysteine residues in the structure, stability, and alkane producing activity of cyanobacterial aldehyde deformylating oxygenase. PLoS One 10, e0122217, https://doi.org/10.1371/journal.pone.0122217 (2015).
Sorigué, D. et al. Microalgae synthesize hydrocarbons from long-chain fatty acids via a light-dependent pathway. Plant Physiol. 171, 2393–405 (2016).
Sorigué, D. et al. An algal photoenzyme converts fatty acids to hydrocarbons. Science 357, 903–907 (2017).
Björn, L. O. Photoenzymes and related topics: an update. Photochem. Photobiol. 94, 459–465 (2018).
Zhang, W. et al. Hydrocarbon synthesis via photoenzymatic decarboxylation of carboxylic acids. J. Am. Chem. Soc. 141, 3116–3120 (2019).
Yunus, I. S. et al. Synthetic metabolic pathways for photobiological conversion of CO2 into hydrocarbon fuel. Metab. Eng. 49, 201–211 (2018).
Voelker, T. A. & Davies, H. M. Alteration of the specificity and regulation of fatty acid synthesis of Escherichia coli by expression of a plant medium-chain acyl-acyl carrier protein thioesterase. J. Bacteriol. 176, 7320–7 (1994).
Steen, E. J. et al. Microbial production of fatty-acid-derived fuels and chemicals from plant biomass. Nature 463, 559–563 (2010).
Burlacot, A. et al. Flavodiiron-mediated O2 photoreduction links H2 production with CO2 fixation during the anaerobic induction of photosynthesis. Plant Physiol. 177, 1639–1649 (2018).
Acknowledgements
This project has received funding from CEA (DRF Impulsion Invention, Alcasun, to F.B.) and from Agence Nationale de la Recherche (PHOTOALKANE, No. ANR-18-CE43-0008-01, to G.P.). This work was also supported by the HelioBiotec platform funded by the EU, the région PACA, the French Ministry of Research, and the CEA. S.M. has received a PhD scholarship from Ecole Normale Supérieure Paris and the French Ministry of Education and Research. Technical support of Stephan Cuiné and Frédéric Espanet (CEA Cadarache) is also acknowledged.
Author information
Authors and Affiliations
Contributions
S.M. performed experiments, analysed the data and wrote the manuscript. B.L., A.B., D.S., S.B. and P.A. assisted in designing experimental set-ups and performing some experiments. F.B. supervised the project and helped write the manuscript. Y.L. and G.P. contributed ideas and corrected the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing Interests
D.S., B.L., S.B., G.P. and F.B. are inventors on an intellectual property application for producing hydrocarbons using methods described in this study.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Moulin, S., Légeret, B., Blangy, S. et al. Continuous photoproduction of hydrocarbon drop-in fuel by microbial cell factories. Sci Rep 9, 13713 (2019). https://doi.org/10.1038/s41598-019-50261-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-50261-6
This article is cited by
-
Photoenzymatic Decarboxylation to Produce Hydrocarbon Fuels: A Critical Review
Molecular Biotechnology (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
