
Abstract
Interferons are potent antiviral cytokines that modulate immunity in response to infection or other danger signals. In addition to their antiviral functions, type I interferons (IFNα and IFNβ) are important in the pathogenesis of autoimmune diseases. Type III interferons (IFNλs) were initially described as a specialized system that inhibits viral replication at epithelial barrier surfaces while limiting inflammatory damage. However, evidence now suggests that type III interferons have complex effects on both innate and adaptive immune responses and might also be pathogenic in systemic autoimmune diseases. Concentrations of IFNλs are increased in blood and tissues in a number of autoimmune rheumatic diseases, including systemic lupus erythematosus, and are further associated with specific clinical and laboratory parameters. This Review is aimed at providing a critical evaluation of the current literature on IFNλ biology and how type III interferons might contribute to immune dysregulation and tissue damage in autoimmunity. The potential effects of type III interferons on treatment strategies for autoimmune rheumatic diseases, such as interferon blockade, are also considered.
Key points
-
Type III interferons (IFNλs) are critical for immune defence against pathogens at epithelial barrier surfaces and were initially described as an anti-inflammatory counterpart to the type I interferon system.
-
IFNλs have complex effects on both innate and adaptive immunity and can promote inflammation in certain contexts.
-
Similar to type I interferons, type III interferon concentrations are increased in the blood and affected tissues of patients with autoimmune rheumatic diseases such as systemic lupus erythematosus (SLE).
-
Concentrations of IFNλs correlate with clinical and immunological parameters and seem to have non-redundant effects on cell-specific and tissue-specific disease processes in SLE.
-
Current biologic therapies that target IFNα or its receptor do not block the effects of IFNλs.
-
Additional research is needed to fully characterize the context-dependent effects of IFNλs and to optimize treatment for patients with autoimmune rheumatic diseases.
Similar content being viewed by others
Introduction
Interferons are a group of cytokines that are produced in response to infection or other inflammatory stimuli. Functionally, these cytokines have potent antiviral effects and modulate immune cell function. Interferons are classified into three subgroups: type I interferons (IFNα, IFNβ, IFNε, IFNκ and IFNω), type II interferon (IFNγ) and type III interferons (four IFNλ subtypes). The type III interferons are a relatively new addition to the interferon family and are especially important in immune defence at barrier surfaces1,2,3. Although type III interferons are structurally distinct from type I interferons, they have overlapping functions, and both signal through the Janus kinase (JAK)–signal transducer and activator of transcription (STAT) pathway to induce transcription of interferon-stimulated genes (ISGs) and promote antiviral activity.
Interferons are critical for host defence, but can also contribute to disease processes in autoimmune and inflammatory diseases. Indeed, dysregulated type I interferon responses are a major feature of systemic lupus erythematosus (SLE) and a number of other systemic autoimmune diseases4. Mutations in genes associated with the type I interferon pathway can also result in monogenic autoinflammatory diseases5,6. Chronic activation of the type I interferon system has myriad effects on both innate and adaptive immune responses. For example, type I interferons can modulate antigen-presenting cell (APC) function, promote B cell activation and antibody production, and induce the production of chemokines that lead to tissue inflammation7. Given the importance of this pathway, biologic agents that target either IFNα or IFNα receptor (IFNAR), the main receptor for type I interferons, have emerged as a potential therapeutic strategy for systemic rheumatic diseases such as SLE8. However, some of these agents have had mixed efficacy in clinical trials, highlighting the complexity and heterogeneity of immune derangement in systemic autoimmunity.
In addition to type I interferons, type III interferons might also contribute to autoimmune and chronic inflammatory diseases. Although type III interferons were initially described as an anti-inflammatory counterpart to the type I interferon system, data suggest that IFNλ biology is more complex than suspected and that excessive and chronic activation of the IFNλ pathway can in fact be detrimental to the host. In this Review, we summarize new insights into IFNλ biology and how type III interferons compare with the type I interferon system. We also discuss potential roles for type III interferons in the immunopathology of systemic rheumatic diseases and explore how this information can be applied to current and future treatment strategies.
IFNλ biology and signalling
Four subtypes of IFNλ have been identified in humans: IFNλ1 (IL-29), IFNλ2 (IL-28A), IFNλ3 (IL-28B) and IFNλ4. Several IFNL pseudogenes are located in the vicinity of the genes encoding IFNλs 1–3 (ref.9), and a common dinucleotide polymorphism in the IFNL locus can result in a frameshift mutation that enables the expression of a functional IFNL4 gene product10. In contrast to humans, only IFNλ2 and IFNλ3 are expressed in mice11.
IFNλs signal through a unique heterodimeric receptor complex comprising IFNλ receptor 1 (IFNLR1) and IL-10 receptor subunit-β12,13. An important difference between type I and type III interferons is the expression of their respective receptor complexes. IFNAR is widely expressed on almost all cell types in the body, whereas expression of the IFNλ receptor (IFNLR) is more limited, being highly expressed on epithelial cells and some immune cells, such as neutrophils in mice and B cells in humans1,2,3. This distribution enables the IFNλ system to have specialized effects at barrier sites.
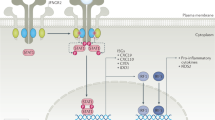
In target cells, the IFNLR complex signals through the JAK–STAT pathway (Fig. 1). IFNα, IFNβ and IFNλs can all activate JAK1 and non-receptor tyrosine-protein kinase TYK2, resulting in the phosphorylation of STAT proteins and the formation of STAT1–STAT2 heterodimers1,2,3. Interferon regulatory factor 9 (IRF9) interacts with these STAT1–STAT2 heterodimers to form the interferon stimulated gene factor 3 (ISGF3) transcription factor complex. ISGF3 then translocates to the nucleus, where it can bind to interferon-stimulated regulatory element sequences located in the promoters of ISGs such as MX1, IFIT1 and ISG15 (ref.14).

Type I and type III interferons can activate both Janus kinase 1 (JAK1) and non-receptor tyrosine-protein kinase TYK2 (TYK2), leading to signal transducer and activator of transcription (STAT) phosphorylation and the formation of STAT1–STAT2 heterodimers. These heterodimers can interact with interferon regulatory factor 9 (IRF9) to form the interferon stimulated gene factor 3 (ISGF3) transcription factor complex. ISGF3 translocates to the nucleus, where it can bind to interferon-stimulated regulatory element (ISRE) sequences and promote the expression of interferon-stimulated genes (ISGs). Type III interferons comparatively induce lower amplitude expression of ISGs over a longer period of time than type I interferons, possibly owing to differential negative regulation by Ubl carboxyl-terminal hydrolase 18 (USP18). Type I and type III interferons can also promote the formation of STAT1 homodimers, which upregulate IRF1 expression and lead to pro-inflammatory chemokine production. IFNλ can also signal through a variety of non-canonical mechanisms. GAS, IFNγ-activated sequence; IFN, interferon; IFNAR, IFNα receptor; IFNLR1, IFNλ receptor 1; IL-10RB, IL-10 receptor subunit-β.
Although type I and type III interferons share downstream signalling machinery, some differences exist in the kinetics of different types of interferon responses. Type III interferons induce longer-lasting expression of ISGs at lower amplitude than type I interferons15,16. This difference might be caused by differential negative regulation by Ubl carboxyl-terminal hydrolase 18, which preferentially inhibits type I interferon signalling but not type III interferon signalling17,18,19. Nevertheless, the transcriptional profiles induced by type I interferons and type III interferons are remarkably similar, and a unique signature for IFNλs has not been identified. Despite these similarities, studies in IFNLR-deficient (Ifnlr1−/−) mice indicate that IFNλs have non-redundant functions in immunity and that type III interferons are particularly important for immune responses at mucosal surfaces20,21,22,23,24,25.
Current thinking suggests that IFNλs restrict viral replication in epithelial cells without inducing inflammatory pathology26. One potential mechanism for the non-inflammatory effects of type III interferons compared with type I interferons is related to chemokine production. IFNβ induces the expression of the chemokines CXCL9, CXCL10 and CXCL11 to a greater extent than IFNλs, owing to insufficient induction of IRF1 by IFNλs27. These chemokines can recruit CXCR3+ leukocytes and are important in the development of tissue inflammation. IFNβ promotes the formation of STAT1 homodimers that bind to the IRF1 promoter and induce IRF1 expression (Fig. 1). By contrast, IFNλs do not induce sufficient IRF1 expression to enable the production of chemokines. Notably, IRF1 induction is dependent on the expression of IFNLR1, as overexpression of IFNLR1 increases the amount of CXC chemokines produced in response to IFNλs to similar levels to those elicited by IFNβ. These findings27 suggest that IFNLR1 density is an important determinant of IFNλ function. As such, IFNλs could theoretically promote inflammation if IFNLR1 expression is sufficiently high to induce IRF1 expression. The way in which IFNLR1 expression is regulated, particularly in autoimmune diseases, might therefore explain the context-dependent effects of IFNλs (discussed in the following sections).
IFNλs can also signal through a variety of non-canonical mechanisms. Data from mouse neutrophils show that IFNλs can activate JAK2 and inhibit reactive oxygen species (ROS) production in a model of intestinal inflammation28. This effect was not mediated through traditional STAT1-dependent signal transduction but rather through JAK2-mediated inhibition of RAC-alpha serine/threonine-protein kinase (AKT). Whether this JAK2–AKT pathway is present and operational in other cell types is currently unclear. IFNλs can also activate the mitogen-activated protein kinase pathway29 and modulate cell–cell tight junctions30, further highlighting the complexity of their biology.
IFNλs in host immunity
IFNλs have direct effects on epithelial cells, inducing a variety of cell-intrinsic mechanisms that restrict viral replication and inhibit viral transmission26. However, evidence indicates that IFNλs have additional functions in orchestrating innate and adaptive immune responses. These functions can be separated into direct effects on IFNλ-responsive cells (Table 1) and indirect effects on non-responsive cell types. The effects of IFNλs on different cell types have been reviewed elsewhere3,31. In this section, we focus specifically on aspects of the IFNλ response axis that are relevant for inflammation and autoimmunity.
Innate immunity
IFNλs have direct effects on various innate immune cell populations (Table 1). Multiple studies report that IFNλs can activate mouse neutrophils to induce STAT1 phosphorylation and ISG expression25,28,32. IFNλs can also increase ROS production by mouse neutrophils, and in vivo experiments show that mice with neutrophil-specific deletion of Ifnlr1 are more susceptible to Aspergillus infection33, indicating that IFNλs regulate antifungal immunity through specific effects on neutrophil function. By contrast, IFNλs can inhibit ROS production and degranulation in mouse neutrophils during intestinal inflammation through a non-translational, STAT1-independent pathway28. Whether human neutrophils are similarly responsive to IFNλs is unclear. Human neutrophils express IFNLR1 and upregulate it in response to inflammatory stimuli such as lipopolysaccharide or fungal infection33. IFNλs can also inhibit TNF-induced ROS production in human neutrophils28 and suppress neutrophil extracellular trap (NET) formation in response to activated platelets or platelet-derived inorganic polyphosphate34. These data are somewhat contradicted by reports that IFNλs do not induce ISG expression in human peripheral blood neutrophils35,36, leading to uncertainty about whether and how these cells respond to IFNλs in different settings.
Dendritic cell (DC) subsets are also an important part of the IFNλ response network. IFNλs can increase type I interferon and chemokine production by human plasmacytoid DCs (pDCs)37,38,39 and can upregulate the expression of class I and II MHC molecules and co-stimulatory molecules on pDCs, which could promote T cell activation37,39. By contrast, IFNλs seem to induce a more regulatory phenotype in human monocyte-derived DCs, which promote the expansion of FOXP3+ regulatory T cells40. Other reports suggest that IFNλs are involved in T cell polarization in vitro41 and that they can skew T cells towards a T helper 1 cell response in a mouse model of allergic asthma through effects on lung DCs41,42. Despite the progress made by these studies, the effects of IFNλs on DC function have not been fully characterized, and it is probable that only certain DC subsets respond directly to this cytokine.
In addition, IFNλs can activate human monocyte-derived macrophages and promote a pro-inflammatory phenotype, leading to chemokine production and the upregulation of pathways related to antigen presentation, co-stimulation, phagocytosis and cytotoxicity43,44,45,46. By contrast, natural killer cells do not seem to respond to IFNλs directly35,36,43,47,48; however, IFNλs can modulate natural killer cell function indirectly through their effects on macrophages43,48,49.
Adaptive immunity
IFNλs also have direct effects on some adaptive immune cells (Table 1). Although IFNλs do not seem to affect mouse B cells and T cells35,50,51,52, data indicate that human lymphocytes can respond to IFNλs. The reasons behind the discrepancies between mouse and human responses remain unclear. Human B cells express IFNLR, and stimulation with IFNλs promotes ISG expression in these cells35,36,53. Moreover, IFNλs increase Toll-like receptor 7 (TLR7)-mediated and TLR8-mediated antibody production and plasmablast differentiation in human B cells54,55. Pre-treatment with IFNλs can also inhibit influenza-induced IgG production in human peripheral blood mononuclear cells (PBMCs)56. However, it is worth noting that this inhibitory effect was observed in a mixed cell population and might result from decreased production of T helper 2 cell cytokines rather than from a direct effect on B cell function. This idea is consistent with other reports that IFNλs promote T helper 1 cell skewing via effects on DCs41,42.
The effects of IFNλs on human T cells are less obvious than the effects on B cells. Several reports indicate that human T cells do not express IFNLR1 and are not responsive to IFNλ stimulation35,53,57. By contrast, a 2020 study has indicated that CD8+ T cells can respond to IFNλs and upregulate ISGs36. Activation of T cells with anti-CD3 and anti-CD28 antibodies also upregulated IFNLR1 on CD4+ T cells, allowing the induction of ISGs by IFNλs36. Therefore, T cells could potentially acquire responsiveness to IFNλs in the context of antigen-specific immune responses.
In addition to direct effects, IFNλs also coordinate adaptive immunity through indirect mechanisms. Ifnlr1−/− mice have decreased antibody and CD8+ T cell responses following infection with influenza virus51. This effect is dependent on thymic stromal lymphopoietin (TSLP) production by microfold cells in the upper airway. IFNλs induce TSLP production in these cells, leading to CD103+ DC migration to the draining lymph nodes. These CD103+ DCs subsequently promote follicular helper T cell expansion and germinal centre responses in the lymph node, thereby generating a robust adaptive immune response. Whether this IFNλ-induced TSLP-mediated mechanism is specific to the respiratory tract and is also present in humans, or whether it can boost adaptive immune responses against self-antigens, remains unclear. A separate study further demonstrated that IFNλs are required for APC migration to the draining lymph nodes and are critical for the development of effective antiviral CD8+ T cell responses during influenza infection in mice58, highlighting another mechanism through which IFNλs can potentiate adaptive immune responses.
In summary, these data suggest that IFNλs can modulate immune responses through a variety of direct and indirect pathways. Although these mechanisms have primarily been identified and studied in response to infection, they might also be relevant for autoimmunity. Considerable differences in the IFNλ response also exist between mouse and human cells. These differences will be important to consider when evaluating IFNλs in the context of human diseases.
IFNλs in autoimmune rheumatic diseases
Concentrations of IFNλs are increased in blood and affected tissues in a number of autoimmune rheumatic diseases, including SLE, rheumatoid arthritis (RA), primary Sjögren syndrome (pSS) and systemic sclerosis (SSc). Increased amounts of IFNλs are also associated with increased disease severity, increased autoantibodies, increased inflammatory markers and/or specific manifestations in these diseases (Table 2). In this section, we summarize the main findings in these diseases and discuss potential mechanisms of immune dysregulation.
Systemic and cutaneous lupus erythematosus
SLE is a complex autoimmune disease that can affect multiple organ systems, including the skin, kidneys, joints and vasculature. The role of type I interferons is well established in SLE pathogenesis, and many patients with SLE display a characteristic type I interferon signature in blood and affected tissues59,60,61. Functionally, type I interferons lead to the aberrant activation of immune cells62 by promoting autoantibody production and immune complex formation that result in tissue damage. Type I interferons can also prime neutrophils, modify APC activity and regulate T cell function to further promote autoimmune tissue damage in SLE.
In addition to type I interferons, evidence suggests that type III interferons are dysregulated in SLE. Several studies have reported that serum IFNλ1 and IFNλ3 concentrations are increased in patients with SLE compared with healthy individuals63,64,65,66,67,68,69,70,71. IFNL1 transcripts are increased in PBMCs and IFNL2 and IFNL3 transcripts are increased in activated CD4+ T cells from patients with SLE relative to those from healthy individuals63,72. Moreover, increased serum concentrations of IFNλs are associated with disease severity and clinical laboratory values. Specifically, higher serum concentrations of IFNλ correlate with higher SLE Disease Activity Index scores63,64,65,66, higher anti-double-stranded DNA (dsDNA) autoantibody titres63,64 and lower amounts of complement proteins C3 and C4 (refs63,64,65,66). Increased circulating concentrations of IFNλs are also associated with the presence of specific disease manifestations in SLE, including arthritis, nephritis, serositis and skin involvement63,65,71. Genetic studies further implicate IFNλs in SLE pathogenesis. IFNL3 and IFNL4 variants are risk factors for lupus nephritis among patients with SLE in a Taiwanese cohort66, and the rs4649203 single nucleotide polymorphism in IFNLR1 is associated with an increased risk of SLE in a Chinese Han population73.
IFNλs have also been detected in affected tissues in patients with SLE. Immunohistochemistry analysis of skin tissue showed that IFNλs and IFNLR1 are substantially increased in patients with chronic discoid lupus erythematosus or subacute cutaneous lupus erythematosus (CLE) relative to healthy individuals or patients with other inflammatory skin diseases (such as atopic dermatitis or psoriasis)69. The detection of IFNλs in the skin of patients with CLE is most prominent in the epidermis, with some additional staining of mononuclear cells in the dermis. Patients with CLE also have increased serum IFNλ1 concentrations, particularly in those patients with disseminated lesions compared with those with more localized disease, and a case report from a single patient found that serum IFNλ1 concentrations declined during clinical remission following treatment with glucocorticoids and hydroxychloroquine69. In addition to skin, IFNλs and IFNLR1 are also detectable in kidney tissue from patients with lupus nephritis66,70; IFNλs were mostly observed in glomerular crescents and areas with inflammatory infiltrates, and glomerular IFNλ staining decreased in repeat biopsy-retrieved samples from patients who achieved a histological response to treatment70. However, these studies did not include kidney tissue samples from healthy individuals or disease-matched controls. Overall, these findings indicate that IFNλs might be involved in SLE-associated skin and kidney disease.
Data from mouse models support a mechanistic role for IFNλs in SLE. One study investigated the effects of IFNλs in a TLR7-induced lupus model, whereby mice are repeatedly exposed to the TLR7 agonist imiquimod. Serum concentrations of IFNλ2 and IFNλ3 were increased in imiquimod-treated mice, and IFNLR1 deficiency substantially reduced splenomegaly and leucocytosis compared with wild-type controls35. Ifnlr1−/− mice were fully responsive to IFNα, suggesting that IFNλs have important and non-redundant functions in systemic immune dysregulation. Further investigation of splenic immune cell populations revealed that IFNλs promote myeloid cell expansion and T cell activation following in vivo TLR7 stimulation, potentially through a combination of direct and indirect effects on these cells. By contrast, IFNλs did not modulate B cell responses in TLR7-induced lupus, as the number of plasma cells and levels of B cell activation markers did not differ between Ifnlr1−/− mice treated with imiquimod and wild-type controls. IFNLR1 deficiency also had no effect on the amounts of circulating antinuclear antibodies or anti-dsDNA autoantibodies. These results35 are somewhat contradictory to data in humans, which indicate that increased concentrations of IFNλs correlate with higher levels of autoantibodies in SLE63,64,67,68. This discrepancy could potentially be related to differences in B cell responsiveness to IFNλs between mice and humans. Mouse B cells are unresponsive to IFNλs, whereas human B cells can respond directly to this cytokine by increasing TLR7-mediated antibody production and plasmablast differentiation54,55. Therefore, additional research is needed to determine how IFNλ affects autoantibody production in the context of human SLE.
Mouse models of lupus also support a role for IFNλs in the pathogenesis of skin and kidney manifestations in SLE. Ifnlr1−/− mice had substantially reduced skin inflammation in the TLR7-induced lupus model35. This decrease in skin inflammation corresponded with a decrease in tissue expression of pro-inflammatory genes such as Il6, Cxcl9 and Cxcl10. In vitro experiments show that mouse and human keratinocytes respond to IFNλs and can upregulate CXCL9, CXCL10 and CXCL11 chemokines35. Moreover, culture supernatants from IFNλ-stimulated human keratinocytes can induce the in vitro migration of mononuclear immune cells35,69. These data suggest that IFNλs might, at least in part, promote SLE-associated skin disease by increasing pro-inflammatory chemokine production in keratinocytes (Fig. 2). Notably, co-treatment of keratinocytes with IFNα and IFNλ1 induced greater chemokine expression than either cytokine alone35, indicating that type I and type III interferons could have an additive effect in promoting skin inflammation. IFNλs also increase the expression of MHC class I molecules by human keratinocytes, which can in turn promote pathogenic CD8+ T cell responses74. In the same TLR7-induced lupus model, Ifnlr1−/− mice also had decreased immune complex deposition, glomerulosclerosis and ISG expression in the kidneys35. IFNλs were able to induce ISGs and chemokine production in mouse mesangial cells, suggesting that IFNλs could have an important effect on structural cells in the kidney. Other kidney cells, in particular those of epithelial origin, can also potentially respond to IFNλs75. Further analysis is required to identify and characterize how IFNλs can affect other tissues, such as the lung, brain and joints, that are commonly involved in SLE; however, unlike type I interferons, type III interferons do not seem to have any effects on vascular disease in mice, as IFNLR1 deficiency did not improve endothelium-dependent vasorelaxation in the TLR7-induced lupus model35.

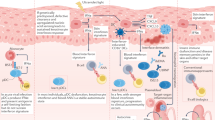
Danger signals, including Toll-like receptor 7 (TLR7) agonists and RNA-containing immune complexes (RNA-IC), can induce the production of IFNλs by plasmacytoid dendritic cells. TLR3 agonists can also induce the production of IFNλs by keratinocytes. IFNλs can subsequently activate keratinocytes to upregulate the expression of the chemokines CXCL9, CXCL10 and CXCL11, as well as surface MHC class I molecules. These chemokines recruit CXCR3+ leukocytes to the skin, where they promote tissue damage; in particular, cytotoxic CD8+ T cells can cause keratinocyte cell death. The release of endogenous nucleic acids (in combination with the cathelicidin LL-37 in experimental models) can induce further production of IFNλs, resulting in a feed-forward loop that perpetuates inflammation in the skin. Whether inflammatory stimuli can also upregulate IFNλ receptor (IFNLR) expression on keratinocytes is unclear. NET, neutrophil extracellular trap.
Interferon production in SLE occurs through a variety of mechanisms involving nucleic acids, immune complexes and the engagement of various intracellular sensors (Fig. 2). pDCs are a major source of type I interferons in SLE76 and are also involved in IFNλ production. These cells accumulate in the skin of mice with lupus and produce IFNλs in response to TLR7 agonists35. In humans, RNA-containing immune complexes can induce the production of type III interferons in a subset of pDCs that also produce type I interferons77. The production of IFNλs by pDCs in vitro was attenuated in the presence of hydroxychloroquine or an IL-1 receptor associated kinase 4 (IRAK4) inhibitor, indicating that RNA-containing immune complexes induce the production of IFNλs through the endosomal TLR–myeloid differentiation primary response protein (MyD88) system. Additional research is needed to determine whether other immune stimuli that trigger type I interferon production, such as NETs78,79, can also contribute to the production of IFNλs in SLE. In addition to pDCs, keratinocytes seem to be a potential source of IFNλs in the skin. Epidermal explants and cultured human keratinocytes produce considerable amounts of IFNλs in response to synthetic TLR3 agonists69. A follow-up study demonstrated that endogenous nucleic acids isolated from keratinocytes, in combination with the cathelicidin LL-37, were able to induce the production of IFNλs80. These results are consistent with the finding that keratinocyte cell death and increased amounts of nuclear debris perpetuate inflammation in SLE skin lesions81 (Fig. 2). Keratinocytes can also upregulate IFNL transcripts after stimulation with IFNα35, highlighting another potential feed-forward pro-inflammatory loop whereby type I interferon amplifies the type III interferon pathway in skin. Overall, current data indicate that IFNλs are potentially pathogenic in SLE, causing cell-specific and tissue-specific effects.
Rheumatoid arthritis
RA is a systemic autoimmune disease that leads to chronic inflammation, cartilage damage and bone erosion in synovial joints. Pro-inflammatory cytokines such as TNF and IL-6 are important in the pathogenesis of RA82; however, blocking these cytokines is not effective in all patients with RA, suggesting that additional pathways are involved. Similar to SLE, a subset of patients with RA display a type I interferon signature in blood83. pDCs and type I interferons have also been detected in RA synovium84,85, further indicating that interferons might contribute to RA immunopathology.
Notably, IFNλs are also upregulated in RA. IFNλ1 is substantially increased in serum from patients with RA compared with serum from healthy individuals or patients with ankylosing spondylitis86,87,88,89. IFNL1 transcripts are also increased in PBMCs from patients with RA and, in addition, IFNλ1 is increased in synovial fluid from patients with RA compared with synovial fluid from patients with osteoarthritis88. Despite there being increased amounts of IFNλs in blood and synovial fluid in RA, data on associations between IFNλs and clinical features in RA are mixed. Several studies have reported no correlations between serum IFNλ1 concentrations and clinical parameters, including the 28-joint Disease Activity Score (DAS28), circulating inflammatory markers (such as C-reactive protein) or RA-associated autoantibodies (rheumatoid factor and anti-citrullinated protein antibodies)86,88. Although IFNλ1 was not associated with the presence of autoantibodies, it was associated with knee joint involvement86. By contrast, a separate study reported that serum IFNλ1 concentrations correlated with the presence of RA-associated autoantibodies, and also correlated with worse DAS28 scores in patients positive for anti-citrullinated protein antibodies89. Moreover, IFNλ1 concentrations decrease following 6 months of treatment with DMARDs89. IFNλ1 concentrations have also been associated with the presence of anti-mutated citrullinated vimentin autoantibodies, and IFNλ2 concentrations are only increased in patients with active RA (defined by a DAS28 score of >2.6)87, suggesting that each IFNλ might contribute to specific disease processes. Further assessment of whether this cytokine modulates human B cell function and autoantibody production in RA will be important.
In the synovium, IFNλ1 co-localizes with CD68+ cells and FGF2+ cells88, suggesting that macrophages and synovial fibroblasts might be relevant sources of IFNλs in RA. In vitro experiments also indicate that synovial fibroblasts can respond to IFNλs. A human RA synovial fibroblast cell line expresses IFNLR1, and stimulation of these cells with recombinant IFNλ1 upregulates IL6, IL8 and MMP3 expression88. IFNλs also upregulate the expression of TLRs 2, 3 and 4 in the same synovial fibroblast cell line, thereby amplifying TLR-mediated IL-6 and IL-8 production90. These results suggest that IFNλs might promote joint inflammation and damage in RA.
Other data indicate that IFNλs actually have the opposite effect and are protective against inflammatory arthritis. In one study, treatment with recombinant IFNλ2 suppressed neutrophil infiltration and IL-1β production in mice with collagen-induced arthritis32. Another study showed that IFNλ1 can inhibit osteoclast formation in vitro91, suggesting that IFNλ1 might be protective against bone erosion in RA. Overall, it is still unclear if IFNλs are pathogenic in RA, and further research is needed to better characterize how IFNλs could contribute to immune dysregulation and joint damage in this disease. Notably, variability in responses to type III interferons by human and mouse cells could complicate interpretation of data in the context of animal models of RA in future studies.
Other autoimmune rheumatic diseases
pSS is a systemic autoimmune condition that targets exocrine glands, resulting in a dry mouth, dry eyes and several systemic manifestations. pSS is characterized by an exaggerated type I interferon response in blood and affected glands92,93, and evidence indicates that IFNλs might also contribute to the immunopathology of pSS. Immunohistochemistry analysis of minor salivary glands has demonstrated that expression of IFNλs is increased in tissue from patients with pSS compared with tissue from individuals with non-pSS sicca symptoms94,95. Serum IFNλ1 concentrations are similarly increased in patients with pSS94.
Salivary gland epithelial cells might contribute to both the production of IFNλs and the IFNλ response in pSS. TLR3 agonists induce the production of IFNλs by salivary gland epithelial cells, which in turn upregulates CXCL10 and BAFF (which encodes B cell activating factor (BAFF)) expression in these cells94,95. Co-treatment with IFNα and IFNλ1 can further enhance STAT1 phosphorylation and cytokine expression in salivary gland epithelial cells, suggesting that type I and type III interferons could have combined effects in pSS. BAFF is a known pathogenic factor in pSS and promotes B cell hyperactivity and autoantibody production92. On the basis of these findings, it will be important to further investigate the potential link between IFNλs and aberrant B cell responses in pSS.
IFNλs also have potential effects in SSc, an autoimmune disease characterized by vasculopathy and widespread fibrosis in the skin, lungs and other organs. The amount of IFNλ1 is increased in the serum of patients with SSc (both diffuse and limited cutaneous subtypes) compared with healthy controls96. Concentrations of IFNλs are highest in patients with SSc who have muscle involvement and correlate positively with concentrations of IFNγ, suggesting that IFNλs might interact with other cytokine networks to amplify pathogenicity in SSc. Notably, the rs12979860 variant of IFNL3 is associated with an increased risk of pulmonary fibrosis in SSc97, whereas no associations have been reported between this variant and skin fibrosis. Serum IFNλ3 concentrations are also higher in patients with SSc who have pulmonary fibrosis than in those patients with SSc who do not develop this complication, and Ifnl3 transcripts are increased in lung tissue in a mouse model of pulmonary fibrosis97. However, additional research is needed to identify the cellular targets and pathways responsible for the pro-fibrotic effects of IFNλs in SSc.
Preliminary evidence also exists suggesting that IFNλ expression is dysregulated in other autoimmune and inflammatory diseases. The expression of IFNλs is increased in skin samples from patients with dermatomyositis69. By contrast, expression of IFNλ1 is decreased in the ocular fluid of patients with juvenile idiopathic arthritis-associated uveitis98. No further investigation has been carried out into how IFNλs might relate to pathogenesis, disease severity or other immunological parameters in these diseases. IFNλs have also been implicated in psoriasis and inflammatory bowel disease99,100, which are beyond the scope of this Review, and it remains unclear if IFNλs are involved in seronegative spondyloarthritis, which is associated with these conditions.
Are IFNλs protective or harmful?
IFNλs seem to have considerable pro-inflammatory and anti-inflammatory effects that are highly context dependent. As discussed in previous sections, concentrations of IFNλs are increased and could have pathogenic effects in autoimmune diseases such as SLE. Data obtained during the COVID-19 pandemic also indicate that persistent IFNλ signalling can disrupt epithelial barrier function in the lungs and predispose individuals to bacterial superinfection101,102, further highlighting the possibility of IFNλ-mediated tissue damage. By contrast, compelling data suggest that IFNλs can be protective against inflammation by regulating neutrophil function in mouse models of arthritis, colitis and thromboinflammation28,32,34, as well as promoting mucosal healing in the gastrointestinal tract100.
One explanation for these seemingly contradictory effects of IFNλs is the expression level of IFNLR. As discussed in a previous section, IFNLR density on epithelial cells seems to regulate the pro-inflammatory effects of IFNλs. Specifically, cells expressing high amounts of IFNLR1 are able to induce sufficient IRF1 expression to produce pro-inflammatory chemokines such as CXCL10 (ref.27). Therefore, it is possible that local or systemic inflammatory processes in SLE and other autoimmune rheumatic diseases can increase IFNLR1 expression above the threshold necessary for IRF1 induction. For example, IFNLR1 staining is increased in the epidermis of patients with CLE69, and Ifnlr1 expression is substantially upregulated in the skin of mice with TLR7-induced lupus compared with healthy controls35 (Fig. 2). Data from primary human hepatocytes suggest that IFNα can upregulate IFNLR1 expression and that this effect is dependent on the IFNL3 genotype of the cells103. Such interactions between type I and type III interferons could also be important in autoimmune rheumatic diseases such as SLE. Additional research is needed to better understand how IFNLR is expressed and regulated in autoimmunity.
Another possibility is that there are cell-specific and tissue-specific differences in IFNLR1 expression, both during homeostasis and in the context of inflammatory pathology. These differences might explain why concentrations of IFNλs correlate with clinical phenotypes in some tissues (such as the skin and kidneys), but not in others. At present, limited data exist on how individual cell types in a tissue respond to IFNλs. Single-cell and spatial transcriptomic analyses will enable better characterization of type III interferon responses in various tissues. Such approaches will help investigators to identify IFNλ-responsive cell types from bulk samples, rather than having to sort individual cell types or use genetically engineered models. Similarly, single-cell approaches will also help researchers to investigate the levels of sensitivity and/or distinct patterns of transcriptional responses to IFNλs in individual cell types or in cells at different stages of differentiation. For example, human neutrophils might gain responsiveness to IFNλs under certain conditions, such as fungal infection33. Although no differences were detected in IFNλ responses between neutrophils from patients with SLE and those from healthy individuals in peripheral blood35, it will be necessary to study leukocyte responses in situ, as these cells could be regulated by local environmental factors in inflamed tissues.
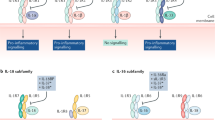
Additionally, IFNλ subtypes could potentially have different immunoregulatory functions. Concentrations of IFNλs 1–3 are all increased in patients with autoimmune rheumatic diseases (Table 2); however, mechanistic studies have largely focused on genetic deletion of Ifnlr1, which abrogates signalling by all IFNλ subtypes. Although there are currently insufficient data to define mechanisms for potential differences between IFNλ subtypes, one possible explanation is their different affinities for IFNLR104. Specifically, IFNλ1 seems to bind to IFNLR with the highest affinity of the IFNλ subtypes, which could generate differences in signalling output, leading to distinct biological potencies of IFNλs105,106. Differential kinetics and magnitudes of IFNλ subtype induction, different stability, bioavailability and tissue distribution of the proteins, and different sensitivity to negative regulators might also result in distinct activities of IFNλ subtypes.
More broadly, IFNλs could have additional roles in immune homeostasis via effects on central tolerance and T cell education in the thymus107. IFNλs are constitutively expressed in medullary thymic epithelial cells and promote MHC class I molecule expression in thymic epithelial cells. IFNλ-induced MHC class I expression seems to be crucial for effective T cell selection, as Ifnlr1−/− mice have impaired negative selection of T cells107. Functionally, this lack of negative selection results in Ifnlr1−/− mice developing spontaneous autoimmune manifestations such as immune cell infiltration in the lung and kidneys. Ifnlr1−/− mice also have increased amounts of total IgG antibodies and develop some tissue-reactive autoantibodies107. Together, these data indicate that IFNλs have myriad effects in different health and disease states.
Implications for treatment
Interferons are important factors underlying the immunopathogenesis of autoimmune rheumatic diseases. Accordingly, the interferon pathway has been an attractive therapeutic target, and several drug candidates (both interferons themselves and interferon-inhibiting therapies) are currently under investigation for SLE and other diseases. Pegylated-IFNλ has been studied as a novel treatment for viral hepatitis and is being tested for COVID-19 (refs108,109,110). Data from mouse models also show that recombinant IFNλ1 suppresses joint inflammation in mice with collagen-induced arthritis by inhibiting neutrophil recruitment32. Accordingly, IFNλ1 has been proposed as a potential treatment for controlling neutrophil-mediated pathology in rheumatic diseases. However, it is still unclear if IFNλ1 has similar effects on human neutrophils to its effects on mouse neutrophils, and it is important to consider off-target effects of IFNλs that might actually worsen autoimmune disease.
The interferon-inhibiting therapies can be categorized into three main groups: drugs that target interferons (both the cytokine and the receptor); drugs that inhibit downstream JAK–STAT signalling; and drugs that inhibit interferon production. Current therapies that target interferons and their receptors only block the type I interferon pathway, whereas therapies that target JAK–STAT signalling components or interferon production can block the effects of both type I and type III interferons (Fig. 3, Table 3).

Biologic agents that target IFNα or IFNα receptor (IFNAR) can block the effects of type I interferons but have no effect on type III interferons. Drugs targeting interferon production by plasmacytoid dendritic cells (such as anti-BDCA2 or anti-LILR4A antibodies), or downstream Janus kinase (JAK)–signal transducer and activator of transcription (STAT) signalling in target cells (such as JAK inhibitors), can block both type I and type III interferons. BDCA2, blood dendritic cell antigen 2; IFN, interferon; IFNGR1, IFNγ receptor 1; IFNGR2, IFNγ receptor 2; IFNLR1, IFNλ receptor 1; IL-10RB, IL-10 receptor subunit-β; LILR4A, leukocyte immunoglobulin-like receptor subfamily A member 4; TYK2, non-receptor tyrosine-protein kinase TYK2.
Targeting interferons
Several neutralizing antibodies that recognize IFNα, including sifalimumab and rontalizumab, have been tested in clinical trials for SLE. A phase II trial of sifalimumab met its primary end point for efficacy of an SLE Responder Index (SRI) response in patients with moderate-to-severe active SLE111. In addition to producing an SRI response, sifalimumab was moderately effective at reducing tissue-specific disease activity in the skin and joints. By contrast, a phase II trial of rontalizumab failed to meet its primary and secondary end points for reducing disease activity in patients with SLE112. The development programmes for both sifalimumab and rontalizumab have since been discontinued, and these therapies are no longer being developed for SLE113. Sifalimumab was also tested in a phase I trial for dermatomyositis and polymyositis, in which it suppressed the interferon gene signature in blood and had some effects in muscle tissue114,115.
An alternative method for blocking IFNα using IFNα kinoid (IFN-K) has also been tested in phase II clinical trials in SLE. In this approach, inactivated IFNα is conjugated to a carrier protein and combined with an adjuvant to induce the production of endogenous anti-IFNα antibodies. Notably, this vaccine-like preparation induces polyclonal antibodies that might be more effective at neutralizing all IFNα subtypes than monoclonal antibodies. Immunization with IFN-K significantly reduced the interferon gene score and met secondary end points for clinical efficacy in patients with SLE116,117,118. However, inducing long-term immunity against interferons could increase the risk of infection119 and the safety profile of IFN-K merits further study.
Overall, antibodies that target IFNα seem to have had mixed efficacy for rheumatic diseases. Because these antibodies specifically target IFNα, it is possible that other type I interferons (such as IFNβ or IFNκ) are still able to bind to the type I interferon receptor without interruption. Moreover, these antibodies have no effect on type III interferons. Of note, a soluble glycoprotein encoded by Yaba-like disease virus can effectively neutralize all human type I and type III interferons120, demonstrating the possibility of developing a pan-interferon antagonist without also targeting signalling components shared by other cytokines (such as occurs with JAK inhibitors). Further research is needed to evaluate the safety and efficacy of this approach.
Targeting interferon receptors
Anifrolumab, a monoclonal antibody that targets IFNAR, is also being investigated in SLE. Data from phase III trials are encouraging, although results are somewhat conflicting. Anifrolumab failed to meet its primary end point of an SRI response in the TULIP-1 trial121. However, anifrolumab significantly reduced disease activity, as measured by a primary BILAG-Based Composite Lupus Assessment response, in the subsequent TULIP-2 trial122. Anifrolumab also reduced glucocorticoid use and improved skin disease in TULIP-2. Further analysis from a phase IIb trial showed that anifrolumab could reduce markers of cardiometabolic dysfunction in patients with SLE, suggesting that it might have additional benefit in SLE vasculopathy123. Anifrolumab is also being evaluated in a phase II trial for patients with active proliferative lupus nephritis124.
In addition to SLE, anifrolumab is also being investigated in other autoimmune rheumatic diseases. Anifrolumab is currently being tested in a phase IIa proof-of-concept trial for patients with moderate-to-severe RA who have an increased interferon gene signature and who have not responded to other biologic DMARDs125. Anifrolumab was also tested in a phase I trial for SSc, in which it suppressed the interferon gene signature in whole blood and skin, which corresponded with a decrease in markers associated with T cell activation and collagen accumulation126,127. However, although targeting IFNAR should block signalling by all type I interferons, it will have no effect on IFNλ signalling. At present, no drugs are available that specifically target IFNλs or their receptor.
Targeting the JAK–STAT signalling pathway
JAK inhibitors are a promising new treatment for SLE that target and block the downstream signalling cascades of multiple cytokines involved in pathogenesis, including both type I and type III interferons. Moreover, these drugs can be given orally as opposed to intravenously like other biologic agents128,129. The JAK1 and JAK2 inhibitor baricitinib met its primary end point for clinical efficacy in a phase II trial in SLE, in which a higher proportion of patients receiving baricitinib achieved resolution of rash or arthritis compared with those who received placebo130. Phase III trials for baricitinib in SLE are ongoing131,132,133. Tofacitinib, a non-selective JAK1 and JAK3 inhibitor, has also shown potential in pre-clinical lupus models121, and in a phase Ib/IIa trial for mild-to-moderate SLE it demonstrated a good safety profile and improved cardiometabolic parameters134,135.
JAK inhibitors have been extensively studied in RA, and several drugs (tofacitinib, baricitinib and upadacitinib) have been approved for clinical use by the FDA (reviewed elsewhere128,129). The efficacy of JAK inhibitors is probably related to their simultaneous targeting of multiple effector cytokines; however, there are currently no data on how JAK inhibitors modulate the interferon response in RA. JAK inhibitors are being investigated for pSS, SSc and myositis. Tofacitinib is currently in a phase I/II trial for pSS136, and a phase I/II trial for early diffuse SSc137 was recently completed, in which the drug was well tolerated and showed trends towards improvement of clinical outcome measures. Tofacitinib was also tested in a proof-of-concept study for refractory dermatomyositis in which it significantly reduced disease activity, as well as serum CXCL9 and CXCL10 concentrations and the interferon gene signature in skin138. Baricitinib is similarly being evaluated in a phase II trial for patients with idiopathic inflammatory myositis139.
Targeting interferon production
pDCs are a primary source of both type I and type III interferons in SLE. Depletion of pDCs in mouse models of lupus attenuates autoimmunity140,141,142, suggesting that targeting these cells might be an effective treatment option. Blood dendritic cell antigen 2 (BDCA2) is expressed specifically on pDCs and is a potent inhibitor of type I and type III interferon induction when ligated39,143. Notably, BDCA2 expression on pDCs from patients with SLE is decreased, and interferon production can be inhibited ex vivo by an anti-BDCA2 monoclonal antibody144. This approach was tested in a phase I trial for SLE. Treatment with BIIB059, an anti-BDCA2 monoclonal antibody, reduced ISG expression in peripheral blood and interferon-induced proteins in active skin lesions145. These findings were associated with improvements in cutaneous disease, as measured by the CLE Disease Area and Severity Index score, and were also associated with reduced CD45+ immune cell infiltration into skin lesions. BIIB059 is still in development and has completed a phase II trial for SLE and CLE for which preliminary results have been announced146.
Other biologic agents that target pDCs are also in development. VIB7734, an anti-leukocyte immunoglobulin-like receptor subfamily A member 4 (LILRA4) monoclonal antibody that targets pDCs for antibody-dependent cellular cytolysis, has completed a phase Ib trial in a variety of autoimmune diseases, including SLE, CLE, pSS, SSc, dermatomyositis and polymyositis147. Preliminary analysis of data in patients with CLE showed that VIB7734 significantly reduced pDCs in blood and skin, which corresponded with a decrease in interferon gene signature and improvement in CLE Disease Area and Severity Index score. Although these studies require further validation and did not discriminate between type I and type III interferons, the efficacy of anti-pDC therapies suggests that targeting upstream pathways could be advantageous over blocking type I interferons alone.
In addition to biologic agents that target BDCA2 or LILRA4, a variety of drugs can inhibit interferon production by other means. Antimalarial drugs such as hydroxychloroquine are commonly used to treat SLE and can inhibit type I and type III interferon production by pDCs in response to TLR7 or TLR9 stimulation77,148. Other TLR inhibitors are currently in various stages of development149.
Conclusions
Type I interferons are central to the immunopathology of rheumatic diseases and are an important target for therapeutic intervention. By contrast, type III interferons are a new addition to the interferon family that have specialized functions, particularly at barrier surfaces. Early reports suggested that unlike type I interferons, type III interferons seemed to limit inflammation and host damage; as such, type III interferons have not been a major focus of research in rheumatology. However, data indicate that type III interferons are not strictly pro-inflammatory or anti-inflammatory; rather, they seem to have context-dependent functions in regulating immune responses. In autoimmune diseases such as SLE, in which concentrations of IFNλs are abnormally elevated and their signalling is chronically activated, IFNλs might promote immune dysregulation and tissue inflammation. In other diseases in which the effects of IFNλs are more tightly regulated, endogenous or exogenously provided IFNλ might have an immunoregulatory function that suppresses inflammation. Although this difference is better understood in the context of infectious disease, improved understanding of the context-dependent functions of IFNλs will be important to optimize treatment and management for patients with autoimmune rheumatic diseases.
References
Lazear, H. M., Nice, T. J. & Diamond, M. S. Interferon-λ: immune functions at barrier surfaces and beyond. Immunity 43, 15–28 (2015).
Wack, A., Terczynska-Dyla, E. & Hartmann, R. Guarding the frontiers: the biology of type III interferons. Nat. Immunol. 16, 802–809 (2015).
Ye, L., Schnepf, D. & Staeheli, P. Interferon-λ orchestrates innate and adaptive mucosal immune responses. Nat. Rev. Immunol. 19, 614–625 (2019).
Crow, M. K., Olferiev, M. & Kirou, K. A. Type I interferons in autoimmune disease. Annu. Rev. Pathol. 14, 369–393 (2019).
Manthiram, K., Zhou, Q., Aksentijevich, I. & Kastner, D. L. The monogenic autoinflammatory diseases define new pathways in human innate immunity and inflammation. Nat. Immunol. 18, 832–842 (2017).
Rodero, M. P. & Crow, Y. J. Type I interferon-mediated monogenic autoinflammation: The type I interferonopathies, a conceptual overview. J. Exp. Med. 213, 2527–2538 (2016).
Ivashkiv, L. B. & Donlin, L. T. Regulation of type I interferon responses. Nat. Rev. Immunol. 14, 36–49 (2014).
Murphy, G. & Isenberg, D. A. New therapies for systemic lupus erythematosus — past imperfect, future tense. Nat. Rev. Rheumatol. 15, 403–412 (2019).
Kotenko, S. V. & Durbin, J. E. Contribution of type III interferons to antiviral immunity: location, location, location. J. Biol. Chem. 292, 7295–7303 (2017).
Prokunina-Olsson, L. et al. A variant upstream of IFNL3 (IL28B) creating a new interferon gene IFNL4 is associated with impaired clearance of hepatitis C virus. Nat. Genet. 45, 164–171 (2013).
Lasfar, A. et al. Characterization of the mouse IFN-lambda ligand-receptor system: IFN-lambdas exhibit antitumor activity against B16 melanoma. Cancer Res. 66, 4468–4477 (2006).
Kotenko, S. V. et al. IFN-lambdas mediate antiviral protection through a distinct class II cytokine receptor complex. Nat. Immunol. 4, 69–77 (2003).
Sheppard, P. et al. IL-28, IL-29 and their class II cytokine receptor IL-28R. Nat. Immunol. 4, 63–68 (2003).
Schneider, W. M., Chevillotte, M. D. & Rice, C. M. Interferon-stimulated genes: a complex web of host defenses. Annu. Rev. Immunol. 32, 513–545 (2014).
Klinkhammer, J. et al. IFN-λ prevents influenza virus spread from the upper airways to the lungs and limits virus transmission. eLife 7, e33354 (2018).
Pervolaraki, K. et al. Differential induction of interferon stimulated genes between type I and type III interferons is independent of interferon receptor abundance. PLoS Pathog. 14, e1007420 (2018).
Blumer, T., Coto-Llerena, M., Duong, F. H. T. & Heim, M. H. SOCS1 is an inducible negative regulator of interferon λ (IFN-λ)-induced gene expression in vivo. J. Biol. Chem. 292, 17928–17938 (2017).
Burkart, C. et al. Usp18 deficient mammary epithelial cells create an antitumour environment driven by hypersensitivity to IFN-λ and elevated secretion of Cxcl10. EMBO Mol. Med. 5, 1035–1050 (2013).
Francois-Newton, V. et al. USP18-based negative feedback control is induced by type I and type III interferons and specifically inactivates interferon alpha response. PLoS ONE 6, e22200 (2011).
Mahlakoiv, T., Hernandez, P., Gronke, K., Diefenbach, A. & Staeheli, P. Leukocyte-derived IFN-α/β and epithelial IFN-λ constitute a compartmentalized mucosal defense system that restricts enteric virus infections. PLoS Pathog. 11, e1004782 (2015).
Nice, T. J. et al. Interferon-λ cures persistent murine norovirus infection in the absence of adaptive immunity. Science 347, 269–273 (2015).
Lin, J. D. et al. Distinct roles of type I and type III Interferons in intestinal immunity to homologous and heterologous rotavirus infections. PLoS Pathog. 12, e1005600 (2016).
Baldridge, M. T. et al. Expression of Ifnlr1 on intestinal epithelial cells is critical to the antiviral effects of interferon lambda against norovirus and reovirus. J. Virol. 91, e02079–16 (2017).
Caine, E. A. et al. Interferon lambda protects the female reproductive tract against Zika virus infection. Nat. Commun. 10, 280 (2019).
Galani, I. E. et al. Interferon-λ mediates non-redundant front-line antiviral protection against influenza virus infection without compromising host fitness. Immunity 46, 875–890.e6 (2017).
Lazear, H. M., Schoggins, J. W. & Diamond, M. S. Shared and distinct functions of type I and type III interferons. Immunity 50, 907–923 (2019).
Forero, A. et al. Differential activation of the transcription factor IRF1 underlies the distinct immune responses elicited by type I and type III interferons. Immunity 51, 451–464.e6 (2019).
Broggi, A., Tan, Y., Granucci, F. & Zanoni, I. IFN-λ suppresses intestinal inflammation by non-translational regulation of neutrophil function. Nat. Immunol. 18, 1084–1093 (2017).
Alase, A. A. et al. IFNλ stimulates MxA production in human dermal fibroblasts via a MAPK-dependent STAT1-independent mechanism. J. Invest. Dermatol. 135, 2935–2943 (2015).
Lazear, H. M. et al. Interferon-λ restricts West Nile virus neuroinvasion by tightening the blood-brain barrier. Sci. Transl Med. 7, 284ra259 (2015).
Zanoni, I., Granucci, F. & Broggi, A. Interferon (IFN)-λ takes the helm: immunomodulatory roles of type III IFNs. Front. Immunol. 8, 1661 (2017).
Blazek, K. et al. IFN-λ resolves inflammation via suppression of neutrophil infiltration and IL-1β production. J. Exp. Med. 212, 845–853 (2015).
Espinosa, V. et al. Type III interferon is a critical regulator of innate antifungal immunity. Sci. Immunol. 2, eaan5357 (2017).
Chrysanthopoulou, A. et al. Interferon lambda1/IL-29 and inorganic polyphosphate are novel regulators of neutrophil-driven thromboinflammation. J. Pathol. 243, 111–122 (2017).
Goel, R. R. et al. Interferon lambda promotes immune dysregulation and tissue inflammation in TLR7-induced lupus. Proc. Natl Acad. Sci. USA 117, 5409–5419 (2020).
Santer, D. M. et al. Differential expression of interferon-lambda receptor 1 splice variants determines the magnitude of the antiviral response induced by interferon-lambda 3 in human immune cells. PLoS Pathog. 16, e1008515 (2020).
Finotti, G., Tamassia, N., Calzetti, F., Fattovich, G. & Cassatella, M. A. Endogenously produced TNF-α contributes to the expression of CXCL10/IP-10 in IFN-λ3-activated plasmacytoid dendritic cells. J. Leukoc. Biol. 99, 107–119 (2016).
Finotti, G., Tamassia, N. & Cassatella, M. A. Synergistic production of TNFα and IFNα by human pDCs incubated with IFNλ3 and IL-3. Cytokine 86, 124–131 (2016).
Yin, Z. et al. Type III IFNs are produced by and stimulate human plasmacytoid dendritic cells. J. Immunol. 189, 2735–2745 (2012).
Mennechet, F. J. & Uze, G. Interferon-lambda-treated dendritic cells specifically induce proliferation of FOXP3-expressing suppressor T cells. Blood 107, 4417–4423 (2006).
Jordan, W. J. et al. Human interferon lambda-1 (IFN-lambda1/IL-29) modulates the Th1/Th2 response. Genes. Immun. 8, 254–261 (2007).
Koltsida, O. et al. IL-28A (IFN-λ2) modulates lung DC function to promote Th1 immune skewing and suppress allergic airway disease. EMBO Mol. Med. 3, 348–361 (2011).
Read, S. A. et al. Macrophage coordination of the interferon lambda immune response. Front. Immunol. 10, 2674 (2019).
Hou, W. et al. Lambda interferon inhibits human immunodeficiency virus type 1 infection of macrophages. J. Virol. 83, 3834–3842 (2009).
Liu, B. S., Janssen, H. L. & Boonstra, A. IL-29 and IFNα differ in their ability to modulate IL-12 production by TLR-activated human macrophages and exhibit differential regulation of the IFNγ receptor expression. Blood 117, 2385–2395 (2011).
Liu, M. Q. et al. IFN-λ3 inhibits HIV infection of macrophages through the JAK-STAT pathway. PLoS ONE 7, e35902 (2012).
Morrison, M. H. et al. IFNL cytokines do not modulate human or murine NK cell functions. Hum. Immunol. 75, 996–1000 (2014).
de Groen, R. A. et al. IFN-λ-mediated IL-12 production in macrophages induces IFN-γ production in human NK cells. Eur. J. Immunol. 45, 250–259 (2015).
Wang, Y. et al. Involvement of NK cells in IL-28B-mediated immunity against influenza virus infection. J. Immunol. 199, 1012–1020 (2017).
Ank, N. et al. An important role for type III interferon (IFN-lambda/IL-28) in TLR-induced antiviral activity. J. Immunol. 180, 2474–2485 (2008).
Ye, L. et al. Interferon-λ enhances adaptive mucosal immunity by boosting release of thymic stromal lymphopoietin. Nat. Immunol. 20, 593–601 (2019).
Misumi, I. & Whitmire, J. K. IFN-λ exerts opposing effects on T cell responses depending on the chronicity of the virus infection. J. Immunol. 192, 3596–3606 (2014).
Kelly, A. et al. Immune cell profiling of IFN-λ response shows pDCs express highest level of IFN-λR1 and are directly responsive via the JAK-STAT pathway. J. Interferon Cytokine Res. 36, 671–680 (2016).
de Groen, R. A., Groothuismink, Z. M., Liu, B. S. & Boonstra, A. IFN-λ is able to augment TLR-mediated activation and subsequent function of primary human B cells. J. Leukoc. Biol. 98, 623–630 (2015).
Syedbasha, M. et al. Interferon-λ enhances the differentiation of naive B cells into plasmablasts via the mTORC1 pathway. Cell Rep. 33, 108211 (2020).
Egli, A. et al. IL-28B is a key regulator of B- and T-cell vaccine responses against influenza. PLoS Pathog. 10, e1004556 (2014).
Jordan, W. J. et al. Modulation of the human cytokine response by interferon lambda-1 (IFN-lambda1/IL-29). Genes. Immun. 8, 13–20 (2007).
Hemann, E. A. et al. Interferon-λ modulates dendritic cells to facilitate T cell immunity during infection with influenza A virus. Nat. Immunol. 20, 1035–1045 (2019).
Baechler, E. C. et al. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc. Natl Acad. Sci. USA 100, 2610–2615 (2003).
Bennett, L. et al. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J. Exp. Med. 197, 711–723 (2003).
Der, E. et al. Tubular cell and keratinocyte single-cell transcriptomics applied to lupus nephritis reveal type I IFN and fibrosis relevant pathways. Nat. Immunol. 20, 915–927 (2019).
Crow, M. K. Type I interferon in the pathogenesis of lupus. J. Immunol. 192, 5459–5468 (2014).
Wu, Q., Yang, Q., Lourenco, E., Sun, H. & Zhang, Y. Interferon-lambda1 induces peripheral blood mononuclear cell-derived chemokines secretion in patients with systemic lupus erythematosus: its correlation with disease activity. Arthritis Res. Ther. 13, R88 (2011).
Adel, Y. & Sadeq, Y. Impact of IL-34, IFN-α and IFN-λ1 on activity of systemic lupus erythematosus in Egyptian patients. Reumatologia 58, 221–230 (2020).
Amezcua-Guerra, L. M. et al. Type III interferons in systemic lupus erythematosus: association between interferon λ3, disease activity, and anti-Ro/SSA antibodies. J. Clin. Rheumatol. 23, 368–375 (2017).
Chen, J. Y. et al. Interferon-λ3/4 genetic variants and interferon-λ3 serum levels are biomarkers of lupus nephritis and disease activity in Taiwanese. Arthritis Res. Ther. 20, 193 (2018).
Oke, V. et al. IFN-λ1 with Th17 axis cytokines and IFN-α define different subsets in systemic lupus erythematosus (SLE). Arthritis Res. Ther. 19, 139 (2017).
Oke, V. et al. High levels of circulating interferons type I, type II and type III associate with distinct clinical features of active systemic lupus erythematosus. Arthritis Res. Ther. 21, 107 (2019).
Zahn, S. et al. Evidence for a pathophysiological role of keratinocyte-derived type III interferon (IFNλ) in cutaneous lupus erythematosus. J. Invest. Dermatol. 131, 133–140 (2011).
Zickert, A. et al. Interferon (IFN)-λ is a potential mediator in lupus nephritis. Lupus Sci. Med. 3, e000170 (2016).
Hu, F. Q. et al. Characterization of autoantibodies and cytokines related to cutaneous lupus erythematosus. Lupus 30, 315–319 (2021).
Lin, S. C., Kuo, C. C., Tsao, J. T. & Lin, L. J. Profiling the expression of interleukin (IL)-28 and IL-28 receptor α in systemic lupus erythematosus patients. Eur. J. Clin. Invest. 42, 61–69 (2012).
Li, Y. et al. Association analyses identifying two common susceptibility loci shared by psoriasis and systemic lupus erythematosus in the Chinese Han population. J. Med. Genet. 50, 812–818 (2013).
Blanco, P. et al. Increase in activated CD8 T lymphocytes expressing perforin and granzyme B correlates with disease activity in patients with systemic lupus erythematosus. Arthritis Rheum. 52, 201–211 (2005).
Sommereyns, C., Paul, S., Staeheli, P. & Michiels, T. IFN-lambda (IFN-lambda) is expressed in a tissue-dependent fashion and primarily acts on epithelial cells in vivo. PLoS Pathog. 4, e1000017 (2008).
Panda, S. K., Kolbeck, R. & Sanjuan, M. A. Plasmacytoid dendritic cells in autoimmunity. Curr. Opin. Immunol. 44, 20–25 (2017).
Hjorton, K., Hagberg, N., Pucholt, P., Eloranta, M. L. & Ronnblom, L. The regulation and pharmacological modulation of immune complex induced type III IFN production by plasmacytoid dendritic cells. Arthritis Res. Ther. 22, 130 (2020).
Goel, R. R. & Kaplan, M. J. Deadliest catch: neutrophil extracellular traps in autoimmunity. Curr. Opin. Rheumatol. 32, 64–70 (2020).
Lood, C. et al. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat. Med. 22, 146–153 (2016).
Scholtissek, B. et al. Immunostimulatory endogenous nucleic acids drive the lesional inflammation in cutaneous lupus erythematosus. J. Invest. Dermatol. 137, 1484–1492 (2017).
Wenzel, J. Cutaneous lupus erythematosus: new insights into pathogenesis and therapeutic strategies. Nat. Rev. Rheumatol. 15, 519–532 (2019).
Guo, Q. et al. Rheumatoid arthritis: pathological mechanisms and modern pharmacologic therapies. Bone Res. 6, 15 (2018).
Lubbers, J. et al. The type I IFN signature as a biomarker of preclinical rheumatoid arthritis. Ann. Rheum. Dis. 72, 776–780 (2013).
Lande, R. et al. Characterization and recruitment of plasmacytoid dendritic cells in synovial fluid and tissue of patients with chronic inflammatory arthritis. J. Immunol. 173, 2815–2824 (2004).
van Holten, J., Smeets, T. J., Blankert, P. & Tak, P. P. Expression of interferon beta in synovial tissue from patients with rheumatoid arthritis: comparison with patients with osteoarthritis and reactive arthritis. Ann. Rheum. Dis. 64, 1780–1782 (2005).
Wu, Q. et al. Serum IFN-λ1 is abnormally elevated in rheumatoid arthritis patients. Autoimmunity 46, 40–43 (2013).
Castillo-Martinez, D. et al. Type-III interferons and rheumatoid arthritis: Correlation between interferon lambda 1 (interleukin 29) and antimutated citrullinated vimentin antibody levels. Autoimmunity 50, 82–85 (2017).
Wang, F. et al. Interleukin-29 modulates proinflammatory cytokine production in synovial inflammation of rheumatoid arthritis. Arthritis Res. Ther. 14, R228 (2012).
Chang, Q. J., Lv, C., Zhao, F., Xu, T. S. & Li, P. Elevated serum levels of interleukin-29 are associated with disease activity in rheumatoid arthritis patients with anti-cyclic citrullinated peptide antibodies. Tohoku J. Exp. Med. 241, 89–95 (2017).
Xu, L. et al. IL-29 enhances Toll-like receptor-mediated IL-6 and IL-8 production by the synovial fibroblasts from rheumatoid arthritis patients. Arthritis Res. Ther. 15, R170 (2013).
Chen, Y. et al. Dendritic cells-derived interferon-λ1 ameliorated inflammatory bone destruction through inhibiting osteoclastogenesis. Cell Death Dis. 11, 414 (2020).
Gottenberg, J. E. et al. Activation of IFN pathways and plasmacytoid dendritic cell recruitment in target organs of primary Sjögren’s syndrome. Proc. Natl Acad. Sci. USA 103, 2770–2775 (2006).
Hjelmervik, T. O., Petersen, K., Jonassen, I., Jonsson, R. & Bolstad, A. I. Gene expression profiling of minor salivary glands clearly distinguishes primary Sjögren’s syndrome patients from healthy control subjects. Arthritis Rheum. 52, 1534–1544 (2005).
Apostolou, E. et al. Expression of type III interferons (IFNλs) and their receptor in Sjögren’s syndrome. Clin. Exp. Immunol. 186, 304–312 (2016).
Ha, Y. J. et al. Increased expression of interferon-λ in minor salivary glands of patients with primary Sjögren’s syndrome and its synergic effect with interferon-alpha on salivary gland epithelial cells. Clin. Exp. Rheumatol. 36, 31–40 (2018).
Dantas, A. T. et al. Interferons and systemic sclerosis: correlation between interferon gamma and interferon-lambda 1 (IL-29). Autoimmunity 48, 429–433 (2015).
Metwally, M. et al. IFNL3 genotype is associated with pulmonary fibrosis in patients with systemic sclerosis. Sci. Rep. 9, 14834 (2019).
Haasnoot, A. M. et al. Ocular fluid analysis in children reveals interleukin-29/interferon-λ1 as a biomarker for juvenile idiopathic arthritis-associated uveitis. Arthritis Rheumatol. 68, 1769–1779 (2016).
Wolk, K. et al. IL-29 is produced by TH17 cells and mediates the cutaneous antiviral competence in psoriasis. Sci. Transl Med. 5, 204ra129 (2013).
Chiriac, M. T. et al. Activation of epithelial signal transducer and activator of transcription 1 by interleukin 28 controls mucosal healing in mice with colitis and is increased in mucosa of patients with inflammatory bowel disease. Gastroenterology 153, 123–138.e8 (2017).
Broggi, A. et al. Type III interferons disrupt the lung epithelial barrier upon viral recognition. Science 369, 706–712 (2020).
Major, J. et al. Type I and III interferons disrupt lung epithelial repair during recovery from viral infection. Science 369, 712–717 (2020).
Duong, F. H. et al. IFN-λ receptor 1 expression is induced in chronic hepatitis C and correlates with the IFN-λ3 genotype and with nonresponsiveness to IFN-α therapies. J. Exp. Med. 211, 857–868 (2014).
Syedbasha, M. et al. An ELISA based binding and competition method to rapidly determine ligand-receptor interactions. J. Vis. Exp. 14, 53575 (2016).
Dellgren, C., Gad, H. H., Hamming, O. J., Melchjorsen, J. & Hartmann, R. Human interferon-lambda3 is a potent member of the type III interferon family. Genes. Immun. 10, 125–131 (2009).
Diegelmann, J. et al. Comparative analysis of the lambda-interferons IL-28A and IL-29 regarding their transcriptome and their antiviral properties against hepatitis C virus. PLoS ONE 5, e15200 (2010).
Benhammadi, M. et al. IFN-λ enhances constitutive expression of MHC class I molecules on thymic epithelial cells. J. Immunol. 205, 1268–1280 (2020).
Dinnon, K. H. III et al. A mouse-adapted model of SARS-CoV-2 to test COVID-19 countermeasures. Nature 586, 560–566 (2020).
Flisiak, R. et al. A randomized study of peginterferon lambda-1a compared to peginterferon alfa-2a in combination with ribavirin and telaprevir in patients with genotype-1 chronic hepatitis C. PLoS ONE 11, e0164563 (2016).
Muir, A. J. et al. A randomized phase 2b study of peginterferon lambda-1a for the treatment of chronic HCV infection. J. Hepatol. 61, 1238–1246 (2014).
Khamashta, M. et al. Sifalimumab, an anti-interferon-α monoclonal antibody, in moderate to severe systemic lupus erythematosus: a randomised, double-blind, placebo-controlled study. Ann. Rheum. Dis. 75, 1909–1916 (2016).
Kalunian, K. C. et al. A phase II study of the efficacy and safety of rontalizumab (rhuMAb interferon-α) in patients with systemic lupus erythematosus (ROSE). Ann. Rheum. Dis. 75, 196–202 (2016).
Isenberg, D. A. & Merrill, J. T. Why, why, why de-lupus (does so badly in clinical trials). Expert. Rev. Clin. Immunol. 12, 95–98 (2016).
Higgs, B. W. et al. A phase 1b clinical trial evaluating sifalimumab, an anti-IFN-α monoclonal antibody, shows target neutralisation of a type I IFN signature in blood of dermatomyositis and polymyositis patients. Ann. Rheum. Dis. 73, 256–262 (2014).
Guo, X. et al. Suppression of soluble T cell-associated proteins by an anti-interferon-α monoclonal antibody in adult patients with dermatomyositis or polymyositis. Rheumatology 53, 686–695 (2014).
Lauwerys, B. R. et al. Down-regulation of interferon signature in systemic lupus erythematosus patients by active immunization with interferon α-kinoid. Arthritis Rheum. 65, 447–456 (2013).
Ducreux, J. et al. Interferon α kinoid induces neutralizing anti-interferon α antibodies that decrease the expression of interferon-induced and B cell activation associated transcripts: analysis of extended follow-up data from the interferon α kinoid phase I/II study. Rheumatology 55, 1901–1905 (2016).
Houssiau, F. A. et al. IFN-α kinoid in systemic lupus erythematosus: results from a phase IIb, randomised, placebo-controlled study. Ann. Rheum. Dis. 79, 347–355 (2020).
Bastard, P. et al. Autoantibodies against type I IFNs in patients with life-threatening COVID-19. Science 370, eabd4585 (2020).
Huang, J. et al. Inhibition of type I and type III interferons by a secreted glycoprotein from Yaba-like disease virus. Proc. Natl Acad. Sci. USA 104, 9822–9827 (2007).
Furie, R. A. et al. Type I interferon inhibitor anifrolumab in active systemic lupus erythematosus (TULIP-1): a randomised, controlled, phase 3 trial. Lancet Rheumatol. 1, e208–e219 (2019).
Morand, E. F. et al. Trial of anifrolumab in active systemic lupus erythematosus. N. Engl. J. Med. 382, 211–221 (2020).
Casey, K. A. et al. Modulation of cardiometabolic disease markers by type I interferon inhibition in systemic lupus erythematosus. Arthritis Rheumatol. 73, 459–471 (2020).
US National Library of Medicine. ClinicalTrials.gov https://www.clinicaltrials.gov/ct2/show/NCT02547922 (2021).
US National Library of Medicine. ClinicalTrials.gov https://www.clinicaltrials.gov/ct2/show/NCT03435601 (2020).
Goldberg, A. et al. Dose-escalation of human anti-interferon-α receptor monoclonal antibody MEDI-546 in subjects with systemic sclerosis: a phase 1, multicenter, open label study. Arthritis Res. Ther. 16, R57 (2014).
Guo, X. et al. Suppression of T cell activation and collagen accumulation by an anti-IFNAR1 mAb, anifrolumab, in adult patients with systemic sclerosis. J. Invest. Dermatol. 135, 2402–2409 (2015).
Gadina, M. et al. Translational and clinical advances in JAK-STAT biology: the present and future of jakinibs. J. Leukoc. Biol. 104, 499–514 (2018).
Schwartz, D. M. et al. JAK inhibition as a therapeutic strategy for immune and inflammatory diseases. Nat. Rev. Drug Discov. 16, 843–862 (2017).
Wallace, D. J. et al. Baricitinib for systemic lupus erythematosus: a double-blind, randomised, placebo-controlled, phase 2 trial. Lancet 392, 222–231 (2018).
US National Library of Medicine. ClinicalTrials.gov https://www.clinicaltrials.gov/ct2/show/NCT03616912 (2021).
US National Library of Medicine. ClinicalTrials.gov https://www.clinicaltrials.gov/ct2/show/NCT03616964 (2021).
US National Library of Medicine. ClinicalTrials.gov https://www.clinicaltrials.gov/ct2/show/NCT03843125 (2021).
Furumoto, Y. et al. Tofacitinib ameliorates murine lupus and its associated vascular dysfunction. Arthritis Rheumatol. 69, 148–160 (2017).
Hasni, S. et al. 183 A phase 1B/2 A trial of tofacitinib, an oral janus kinase inhibitor, in systemic lupus erythematosus [abstract]. Lupus Sci. Med. 6, A139 (2019).
US National Library of Medicine. ClinicalTrials.gov https://www.clinicaltrials.gov/ct2/show/NCT04496960 (2021).
US National Library of Medicine. ClinicalTrials.gov https://www.clinicaltrials.gov/ct2/show/NCT03274076 (2020).
Paik, J. J. et al. Study of tofacitinib in refractory dermatomyositis (STIR): an open label pilot study of 10 patients. Arthritis Rheumatol. https://doi.org/10.1002/art.41602 (2020).
US National Library of Medicine. ClinicalTrials.gov https://www.clinicaltrials.gov/ct2/show/NCT04208464 (2019).
Rowland, S. L. et al. Early, transient depletion of plasmacytoid dendritic cells ameliorates autoimmunity in a lupus model. J. Exp. Med. 211, 1977–1991 (2014).
Sisirak, V. et al. Genetic evidence for the role of plasmacytoid dendritic cells in systemic lupus erythematosus. J. Exp. Med. 211, 1969–1976 (2014).
Yokogawa, M. et al. Epicutaneous application of Toll-like receptor 7 agonists leads to systemic autoimmunity in wild-type mice: a new model of systemic lupus erythematosus. Arthritis Rheumatol. 66, 694–706 (2014).
Dzionek, A. et al. BDCA-2, a novel plasmacytoid dendritic cell-specific type II C-type lectin, mediates antigen capture and is a potent inhibitor of interferon alpha/beta induction. J. Exp. Med. 194, 1823–1834 (2001).
Blomberg, S., Eloranta, M. L., Magnusson, M., Alm, G. V. & Ronnblom, L. Expression of the markers BDCA-2 and BDCA-4 and production of interferon-alpha by plasmacytoid dendritic cells in systemic lupus erythematosus. Arthritis Rheum. 48, 2524–2532 (2003).
Furie, R. et al. Monoclonal antibody targeting BDCA2 ameliorates skin lesions in systemic lupus erythematosus. J. Clin. Invest. 129, 1359–1371 (2019).
Werth, V. et al. OP0193 BIIB059, a humanized monoclonal antibody targeting BDCA2 on plasmacytoid dendritic cells (pDC), shows dose-related efficacy in the phase 2 LILAC study in patients (pts) with active cutaneous lupus erythematosus (CLE) [abstract]. Ann. Rheum. Dis. 79, 120–121 (2020).
US National Library of Medicine. ClinicalTrials.gov https://www.clinicaltrials.gov/ct2/show/NCT03817424 (2020).
Schrezenmeier, E. & Dorner, T. Mechanisms of action of hydroxychloroquine and chloroquine: implications for rheumatology. Nat. Rev. Rheumatol. 16, 155–166 (2020).
Smith, N. et al. Control of TLR7-mediated type I IFN signaling in pDCs through CXCR4 engagement — a new target for lupus treatment. Sci. Adv. 5, eaav9019 (2019).
Selvakumar, T. A. et al. Identification of a predominantly interferon-λ-induced transcriptional profile in murine intestinal epithelial cells. Front. Immunol. 8, 1302 (2017).
US National Library of Medicine. ClinicalTrials.gov https://www.clinicaltrials.gov/ct2/show/NCT02794285 (2021).
Fleischmann, R. et al. Placebo-controlled trial of tofacitinib monotherapy in rheumatoid arthritis. N. Engl. J. Med. 367, 495–507 (2012).
van Vollenhoven, R. F. et al. Tofacitinib or adalimumab versus placebo in rheumatoid arthritis. N. Engl. J. Med. 367, 508–519 (2012).
Lee, E. B. et al. Tofacitinib versus methotrexate in rheumatoid arthritis. N. Engl. J. Med. 370, 2377–2386 (2014).
Fleischmann, R. et al. Efficacy and safety of tofacitinib monotherapy, tofacitinib with methotrexate, and adalimumab with methotrexate in patients with rheumatoid arthritis (ORAL Strategy): a phase 3b/4, double-blind, head-to-head, randomised controlled trial. Lancet 390, 457–468 (2017).
Genovese, M. C. et al. Baricitinib in patients with refractory rheumatoid arthritis. N. Engl. J. Med. 374, 1243–1252 (2016).
Taylor, P. C. et al. Baricitinib versus placebo or adalimumab in rheumatoid arthritis. N. Engl. J. Med. 376, 652–662 (2017).
Fleischmann, R. et al. Baricitinib, methotrexate, or combination in patients with rheumatoid arthritis and no or limited prior disease-modifying antirheumatic drug treatment. Arthritis Rheumatol. 69, 506–517 (2017).
US National Library of Medicine. ClinicalTrials.gov https://www.clinicaltrials.gov/ct2/show/NCT03978520 (2021).
Rubbert-Roth, A. et al. Trial of upadacitinib or abatacept in rheumatoid arthritis. N. Engl. J. Med. 383, 1511–1521 (2020).
Burmester, G. R. et al. Safety and efficacy of upadacitinib in patients with rheumatoid arthritis and inadequate response to conventional synthetic disease-modifying anti-rheumatic drugs (SELECT-NEXT): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet 391, 2503–2512 (2018).
Genovese, M. C. et al. Safety and efficacy of upadacitinib in patients with active rheumatoid arthritis refractory to biologic disease-modifying anti-rheumatic drugs (SELECT-BEYOND): a double-blind, randomised controlled phase 3 trial. Lancet 391, 2513–2524 (2018).
Smolen, J. S. et al. Upadacitinib as monotherapy in patients with active rheumatoid arthritis and inadequate response to methotrexate (SELECT-MONOTHERAPY): a randomised, placebo-controlled, double-blind phase 3 study. Lancet 393, 2303–2311 (2019).
Acknowledgements
The work of R.R.G. and M.J.K. was supported by the Intramural Research Program at the National Institute of Arthritis and Musculoskeletal and Skin Diseases and the US National Institutes of Health.
Author information
Authors and Affiliations
Contributions
R.R.G. and M.J.K. researched data for the article. All authors contributed substantially to discussion of the content. R.R.G. and M.J.K. wrote the article. All authors reviewed and/or edited the manuscript before submission.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests. The National Institute of Arthritis and Musculoskeletal and Skin Diseases has collaborative research agreements with Medimmune/AstraZeneca and Pfizer that pertain to anti-interferon therapies and Janus kinase inhibitors, respectively.
Additional information
Peer review information
Nature Reviews Rheumatology thanks J.-Y. Chen and R. Hartmann for their contribution to the peer review of this work.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Goel, R.R., Kotenko, S.V. & Kaplan, M.J. Interferon lambda in inflammation and autoimmune rheumatic diseases. Nat Rev Rheumatol 17, 349–362 (2021). https://doi.org/10.1038/s41584-021-00606-1
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41584-021-00606-1
This article is cited by
-
Strategies to therapeutically modulate cytokine action
Nature Reviews Drug Discovery (2023)
-
Are comorbidities associated with the cytokine/chemokine profile of persistent apical periodontitis?
Clinical Oral Investigations (2023)
-
Potential relevance of type I interferon-related biomarkers for the management of polygenic autoimmune rheumatic diseases with childhood onset
Clinical Rheumatology (2023)
