
Abstract
Ferritin is a protein (ca. 12 nm) with a central pocket of 6 nm diameter, and hydrated iron oxide stored in this central cavity of this protein. The protein shell has a complicated structure with 24 subunits. Transmission electron microscopy images of ferritin showed nanosized iron oxides (ca. 4–6 nm) in the protein structure. In high-resolution transmission electron microscopy images of the iron core, d-spacings of 2.5–2.6 Å were observed, which is corresponded to d-spacings of ferrihydrite crystal structure. Our experiments showed that at pH 11, the modified electrode by this biomolecule is active for water oxidation (turnover frequency: 0.001 s−1 at 1.7 V). Using affected by bacteria, we showed that Fe ions in the structure of ferritin are critical for water oxidation.
Similar content being viewed by others

Introduction
Hydrogen production by water splitting is an interesting and promising strategy to store of sustainable but intermittent energies1,2,3. Although the cathodic reaction is of major importance in water splitting, water oxidation in the anode is a limitation for water splitting in according to large overpotential and stability issues4,5,6,7,8,9. Ruthenium and iridium-based catalysts show efficient activity for water oxidation10,11, but these catalysts are scarce and expensive, which is a serious drawback if the catalysts are to be used on a large scale. Earth-abundant elements-based catalysts should be considered for large-scale deployment. Iron is low-cost, environmentally friendly and high availability. Thus, iron-based water oxidizing catalysts are optimistic about being used in water–splitting systems12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55.
Although oxidation of water by FeCl3 was suggested thermally and photochemically many years ago37, the first report on water oxidation by iron compounds in the presence of an oxidant was reported by Elizarova et al. in 198136. The research group observed water oxidation by FeCl3 and [Fe2O(Phen)4]Cl4, (Phen: 1,10-Phenanthroline), in the presence of [Ru(bpy)3]3+. During the reaction pH changes from 10 to 4.5. It is important to note that iron(II) and (III) ions are labile56 in metal complex and iron oxide is formed at pH ≥ 7. Thus under the condition of Elizarova’s reaction, iron oxide could be a true catalyst for water oxidation. Although, the stability of [Ru(bpy)3]3+ at pH 7 should also be checked because even low amounts of RuO2 from the decomposition of [Ru(bpy)3]3+ could efficiently catalyze water oxidation. Parmon et al. used Fe(OH)3 in the presence of [Ru(bpy)3]3+ at pH 10-11 and showed high activity toward water oxidation38. The used Fe(OH)3 was prepared by conventional procedures39, and it may not be optimized for water oxidation. Iron oxides with a higher activity for water oxidation could be prepared by other methods. Especially, now it is found that nanoparticles of iron oxides show efficient activity for water oxidation57. However, one investigation indicated that iron oxide is not formed and unlike to be a catalyst under acidic conditions18. Other investigations show that different phases of iron oxides are usually catalysts for water oxidation under non-acidic conditions12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55. Although under acidic conditions and in the presence of an iron complex, iron oxide is not a true catalyst18, FeO42− oxidizes water in acidic condition, but it could not perform water oxidation catalytically40.
In 2010, Bernhard and Collins reported a Fe-centered tetra amido macrocyclic ligand that efficiently catalyzes water oxidation (turnover frequency (TOF): 1.3 s−1), but a low turnover number ((TON) = 16) in the presence of a chemical oxidant (cerium ammonium nitrate (CAN)) was observed for the complex58. In 2011, Fe complexes with N-donor ligands were reported that efficiently catalyzed water oxidation with high efficiency for hours (turnover numbers more than 350 at pH = 1 and more than 1000 at pH = 214. Using different methods, it was indicated that the Fe complex is a the true catalyst for water oxidation.
The catalytic activity of a few Fe complexes with N-donor ligands was investigated towards water oxidation at pH = 7–9. The study was performed by chemical methods, using [Ru(bpy)3](ClO4)3 (bpy = bipyridine) as an oxidant and showed the simple uncomplexed Fe(III) ion is even more active than the other Fe complexes under these conditions16. In the presence of [Ru(bpy)3](ClO4)3, the group proposed that Fe complexes are decomposed under oxidizing conditions to metal oxides, which form the true catalysts for water oxidation16. The Fe oxide particles formed in the water-oxidation conditions were isolated and detected to be Fe2O3 by the use of various techniques16. The results Fe complexes in the presence of Ce(IV) and [Ru(bpy)3](ClO4)3 suggest that the active Fe catalysts for water oxidation at acidic and basic conditions are different16. At acidic conditions, water oxidation by the Fe complexes appears to go through a molecular-based intermediate, with no Fe oxide formation. On the other hand, at basic conditions, Fe2O3 is the true catalyst for water oxidation16.
For water oxidation by some iron complexes in the presence of cerium(IV), Codola’ et al. reported a FeIV–O–CeIV adduct, which showed cerium(IV) is not a simple and an innocent one-electron oxidant during the water oxidation reaction12.
Kottrup and Hetterscheid studied an on-line mass spectrometry approach to determine the onset of water oxidation and to determine between competing reactions oxygen evolution and carbon dioxide formation28. However, it is important to note that decomposition necessarily does not form carbon dioxide. Carbon dioxide is the product of deep oxidation of the ligand. Thus, decoordination of the ligand from complex could not be detected on-line mass spectrometry28.
Under acidic condition, the nature of α-[Fe(OTf)2(mcp)] (mcp = N,N′-dimethyl-N,N′-bis(pyridin-2- ylmethyl)cyclohexane-1,2-diamine, OTf = trifluoromethanesulfonate anion), under water oxidation and in the presence of cerium(IV) was investigated. Mössbauer spectroscopy showed that the complex produces oxo iron(IV)31. Using HPLC, the decomposition of the catalysts have been studied by identifying ligand fragments that form upon decomposition31. This analysis corresponds to the water oxidation activity of this catalyst with stability against degradation of the ligand via aliphatic C-H oxidation. This finding has served for the synthesis of a catalyst where sensitive C-H bonds have been replaced by C-D bonds31. Deuterated analog, displayed substantially more robust towards oxidative degradation and yields more than 3400 turnover frequency (TON). The information provides evidence that the water oxidation by a molecular structure than iron oxides31. Thus for many iron-based complexes, at low pH values, the true catalyst has molecular-based and in higher pH true catalyst has iron (hydr)oxide-based structures. It seems that to synthesize a stable iron complex under water-oxidation condition at higher pH (≥6), the stability of both ligand and complex is important. The stability of the ligand is not enough because even a stable ligand does not grantee the stability of the complex because the decoordination of the ligand from metal ion without degradation of the ligand is possible31.
In 2016, Masaoka’s group reported a pentanuclear iron complex with 3,5-bis(2-pyridyl)pyrazole ligand, which catalysis water oxidation with a turnover frequency of 1,900 per second52. In the structure, five iron ions are ligated by six dinucleating ligands, where three metal ions are also strongly electronically coupled via an oxo-bridge52. In acetonitrile, the complex exhibits four reversible one-electron oxidation-reduction steps corresponding to Fe(II)/Fe(III) couples, as shown by cyclic voltammetry. When water was added to the acetonitrile, a large electrocatalytic wave was seen at the potential corresponding to the fourth one-electron oxidation process, which is related to the catalytic water oxidation52. The research group proposed that the pentacoordinate equatorial iron centers of the complex provide two adjacent sites available for water binding and activation, which leads to pre-organized O-O bond formation52.
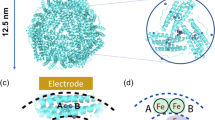
As it was noted, iron oxides are an important iron-based catalyst for water oxidation. Ferritin (Fig. 1), the most important molecule participated in iron storage, storages non-heme iron. Ferritin is a globular protein (12 nm in diameter) with a central cavity (6 nm diameter)59,60,61,62. Iron is stored as hydrated iron oxide in the central cavity. The protein shell has a complicated structure with 24 subunits59,60,61,62.

Nucleation sites (top) and schematic representation of the formation of the first iron clusters. Ferritin core growth (bottom): from the nucleation sites to the final iron oxide nanoparticle. The image and caption were from ref.59. Copyright (2013) by Elsevier.
Results
Transmission electron microscopy (TEM)) images of ferritin showed nanosized iron oxides (ca. 4–6 nm) in the protein structure.
An iron-based core is not significantly larger than 8 nm independent of the different morphologies, as previously reported implies that the ferritin cavities have an inner diameter no greater than 8 nm59,60,61,62.
High-resolution transmission electron microscopy (HRTEM) image of the iron core crystal structure indicates d-spacings of 2.5–2.6 Å, which is corresponded to d-spacings of ferrihydrite crystal structure (Fig. 2)59,60,61,62,63. Selected area (electron) diffraction (SAED), as crystallographic experimental methods, indicates diffuse rings attributed to a low crystalline compound.

(HR)TEM images (a–d) of ferritin from the equine spleen (Scale bar for the image a is 10 nm; scale bar for the image b–d are 5 nm). SAED (e: scale bare: 1/5 nm) showed that the compound has a low crystalline. Red particles show the iron oxide section of ferritin. The black arrows show nanosized iron oxide. The red lines indicate d-spacings of 2.5–2.6 Å, which is corresponded to d-spacings of ferrihydrite crystal structure. Image a and b are colored forms obtained with the ImageJ software. We thank the ImageJ group (W. S. Rasband, ImageJ, U. S. National Institutes of Health, Bethesda, Maryland, USA, http://imagej.nih.gov/ij/, 1997–2016) for the use of the software. Schematic image shows a proposed mechanism for water oxidation by ferrihydrite in the structure of Ferritin.
A proposed mechanism for water oxidation in the presence of ferrihydrite of ferritin is shown in Fig. 2.
The cyclic voltammetry (CV) for ferritin was performed at pH = 11 (Fig. 3a,b; all potentials are reported vs. the normal hydrogen electrode (NHE)). No especial peak was observed for ferritin even at different concentrations (Fig. 3a,b).

CVs for ferritin (red; 3.0 μM) and a bare FTO (black) (a) in the mixture of phosphate buffer (0.25 M, pH = 11.0) and scan rate 10.0 mV/s. CVs for different amounts of ferritin (b) in the mixture of phosphate buffer (0.25 M, pH = 11.0) and scan rate 100.0 mV/s. LSV for ferritin (red; 3 μM) and a fresh FTO (c) in the mixture of phosphate buffer (0.25 M, pH = 11.0) and scan rate 10.0 mV/s. SWV (amplitude: 100 mV; frequency: 10 Hz) for different amounts of ferritin (d) in the mixture of phosphate buffer (0.25 M, pH = 11.0). CV for ferritin at different scan rates in the mixture of phosphate buffer (0.25 M, pH = 11.0) (e). The plot of j vs. scan rate (f). Ferritin was placed on the FTO by Nafion. 3.0 μL or the reported amount of ferritin was dripped on the surface of an FTO and dried at room temperature, then 1.6 μL of 0.5 wt % Nafion solution was placed onto the surface of the electrode.
As shown in Fig. 3c, ferritin is active for water oxidation compared to a bare FTO. Linear sweep voltammetry (LSV) shows the onset of the water-oxidation reaction as 1.46 V and 1.60 V for ferritin and a bare FTO, respectively (Fig. 3c). Thus, LSV shows that the onset for water oxidation is 140 mV less for FTO in the presence of ferritin than the bare FTO. No peak was also indicated for ferritin in CV and LSV. It is not surprising because iron oxide shows no peak under this condition. LSV shows that at 1.70 V and using FTO electrode, the water oxidation is four times more in the presence of ferritin.
Square wave voltammetry, a useful method to detect details, showed no peak for ferritin (Fig. 3d). The effect of scan rates is shown in Fig. 3e,f. At higher scan rates (>1 V/s), the current density is constant (Fig. 3e,f).
In the next step, we tested oxygen evolution by ferritin. As shown in Fig. 4a,b, oxygen evolution was observed for ferritin.

The amperometry (a) and oxygen evolution (b) for fresh ferritin (dot line) and a bare FTO (dash line) and subtraction of FTO on ferritin (solid line) at 1.7 V. The amperometry (c) and oxygen evolution (d) for ferritin effected by bacteria (dot line) and a bare FTO (dash line) subtraction of FTO from ferritin (solid line) at 1.7 V. Ferritin was placed on the FTO by Nafion. 3.0 μL of ferritin was dripped onto the FTO electrode surface and dried at room temperature, then 1.6 μL of 0.5 wt % Nafion solution was placed onto the surface of the electrode in the mixture of phosphate buffer (0.25 M, pH = 11.0) .
Assuming all iron ions are active for water oxidation, a turnover frequency of 0.001 s−1 at 1.7 V was calculated. FTO also indicated oxygen evolution under these conditions.
Streptococcus is proposed to be able to directly extract iron from ferritin, transferrin, lactoferrin, and hemoproteins, or indirectly by small siderophore molecules64. Siderophores (from the Greek: “iron carriers”) are relatively small molecules, which specifically chelates Fe(III) ions. These molecules are synthesized by bacteria and fungi under low iron stress65. Interestingly, the contaminated ferritin by bacteria (Staphylococcus64 and Streptococcus66 bacteria) shows lower activity than fresh ferritin, in fact, iron was consumed by the bacteria64,66, and evolution of oxygen was decreased due to the consumption (Fig. 4c,d).
It seems that in the absence of iron oxide, the decomposition of protein occurs under the harsh condition of water oxidation (Fig. 4c).
Discussion
Recently, iron-based films have been investigated as electrocatalysts for water oxidation, especially Fe2O3 have been reported to be photocatalysts for water oxidation. Wu et al. reported iron oxides as a highly efficient catalyst for water oxidation55. The research group synthesized the catalyst on the surface of the electrodes from Fe(II) ions using simple cyclic voltammetry. As iron oxides are usually insulation, only a thin layer of iron oxide is an efficient catalyst for water oxidation55. An important finding is that the extremely low iron oxide is loading on the surface of the electrodes. Similarly, low amounts of iron oxide could be provided by ferritin for water oxidation55.
Water oxidation by metal oxides usually was reported at pH 13 or 14. However, a comparison for the modified electrode by ferritin at pH 11 by other metal oxides was indicated in Table S1.
The goal of this study is trying to overwhelm the limitations of natural and synthetic materials using a hybrid such as FTO/ferritin. Thus, it is a first step toward finding if a biomolecule such as ferritin could interface with FTO as a synthetic material toward the synthesize a semi-artificial electrode or not?2 Under our electrochemical conditions, the distance between iron oxides, which is placed in the enzyme’s pocket and the surface of the electrode causes a difficult way to the electron transfer. According to the location of the iron oxide in the chiral pocket of ferritin, such system is interesting to be used and developed efficient catalysts to oxidize and reduce both organic and inorganic substrates with a high selectively. For ferritin, at least a small size of iron oxide (6–8 nm) in a hydrophilicity/hydrophobicity pocket is interesting for a first-step investigation.
In addition to different strategies, which use iron-based or non-iron-based catalysts for water oxidation12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,67,68,69,70,71,72,73,74,75, using a bimolecular structure is also interesting and could be a map road to future engineering artificial enzymes for water oxidation or other necessary reactions. Requirements for such hybrids are an increase in the compatibility of the electrode/enzyme, high porosity of the electrode, improvement in the electrode/enzyme interaction.
In summary, water-oxidation activity of ferritin from equine spleen was investigated. The biomolecule can be considered as nanosized iron oxides in a polymer matrix. The experiments showed that at pH 11, the compound is active for water oxidation with a turnover frequency of 0.001 s−1 at 1.7 V.
References
Lewis, N. S. & Nocera, D. G. Powering the planet: Chemical challenges in solar energy utilization. Proc. Natl. Acad. Sci. USA 103, 15729–15735 (2006).
Kornienko, N., Zhang, J. Z., Sakimoto, K. K., Yang, P. & Reisner, E. Interfacing nature’s catalytic machinery with synthetic materials for semi-artificial photosynthesis. Nat. Nanotechnol. 13, 890 (2018).
Ong, W. J., Tan, L. L., Ng, Y. H., Yong, S. T. & Chai, S. P. Graphitic carbon nitride (g-C3N4)-based photocatalysts for artificial photosynthesis and environmental remediation: are we a step closer to achieving sustainability? Chem. Rev. 116, 7159–7329 (2016).
Rüttinger, W. & Dismukes, G. C. Synthetic water-oxidation catalysts for artificial photosynthetic water oxidation. Chem. Rev. 97, 1–24 (1997).
Yagi, M. & Kaneko, M. Molecular catalysts for water oxidation. Chem. Rev. 101, 21–36 (2001).
Najafpour, M. M. et al. Manganese compounds as water-oxidizing catalysts: from the natural water-oxidizing complex to nanosized manganese oxide structures. Chem. Rev. 116, 2886–2936 (2016).
Blakemore, J. D., Crabtree, R. H. & Brudvig, G. W. Molecular catalysts for water oxidation. Chem. Rev. 115, 12974–3005 (2015).
Hunter, B. M., Gray, H. B. & Muller, A. M. Earth-abundant heterogeneous water oxidation catalysts. Chem. Rev. 116, 14120–14136 (2016).
Karkas, M. D., Verho, O., Johnston, E. V. & Åkermark, B. Artificial photosynthesis: molecular systems for catalytic water oxidation. Chem. Rev. 114, 11863–12001 (2014).
Duan, L. et al. A molecular ruthenium catalyst with water-oxidation activity comparable to that of photosystem II. Nat. Chem. 4, 418 (2012).
Moore, G. F. et al. A visible light water-splitting cell with a photoanode formed by codeposition of a high-potential porphyrin and an iridium water-oxidation catalyst. Eng. Environ. Sci. 4, 2389–2392 (2011).
Codolà, Z. et al. Evidence for an oxygen evolving iron–oxo–cerium intermediate in iron-catalysed water oxidation. Nat. comm. 6, 5865 (2015).
Codolà, Z. et al. Electronic effects on single-site iron catalysts for water oxidation. Chem–A. Eur. J. 19, 8042–8047 (2013).
Fillol, J. L. et al. Efficient water oxidation catalysts based on readily available iron coordination complexes. Nat. Chem. 3, 807 (2011).
Tan, P., Kwong, H. K. & Lau, T. C. Catalytic oxidation of water and alcohols by a robust iron (III) complex bearing a cross-bridged cyclam ligand. Chem. Comm. 51, 12189–12192 (2015).
Chen, G., Chen, L., Ng, S. M., Man, W. L. & Lau, T. C. Chemical and visible‐light‐driven water oxidation by iron complexes at ph 7–9: Evidence for dual‐active intermediates in iron‐catalyzed water oxidation. Angew. Chem. Int. Ed. 52, 1789–1791 (2013).
To, W. P. et al. Water oxidation catalysed by iron complex of N, N′-dimethyl-2, 11-diaza [3, 3](2, 6) pyridinophane. Spectroscopy of iron–oxo intermediates and density functional theory calculations. Chem. Sci. 6, 5891–5903 (2015).
Hong, D. et al. Water oxidation catalysis with nonheme iron complexes under acidic and basic conditions: homogeneous or heterogeneous? Inorg. Chem. 52, 9522–9531 (2013).
Das, B. et al. Water oxidation catalyzed by molecular di-and nonanuclear Fe complexes: importance of a proper ligand framework. Dalton Trans. 45, 13289–13293 (2016).
Wickramasinghe, L. D. et al. Iron complexes of square planar tetradentate polypyridyl-type ligands as catalysts for water oxidation. J. Am. Chem. Soc. 137, 13260–13263 (2015).
Coggins, M. K., Zhang, M. T., Vannucci, A. K., Dares, C. J. & Meyer, T. J. Electrocatalytic water oxidation by a monomeric amidate-ligated Fe (III)–aqua complex. J. Am. Chem. Soc. 136, 5531–5534 (2014).
Liu, Y., Xiang, R., Du, X., Ding, Y. & Ma, B. An efficient oxygen evolving catalyst based on a μ-O diiron coordination complex. Chem. Commun. 50, 12779–12782 (2014).
Klepser, B. M. & Bartlett, B. M. Anchoring a molecular iron catalyst to solar-responsive WO3 improves the rate and selectivity of photoelectrochemical water oxidation. J. Am. Chem. Soc. 136, 1694–1697 (2014).
Panchbhai, G., Singh, W. M., Das, B., Jane, R. T. & Thapper, A. Mononuclear iron complexes with tetraazadentate ligands as water oxidation catalysts. Eur. J. Inorg. Chem. 2016, 3262–3268 (2016).
Najafpour, M. M., Safdari, R., Ebrahimi, F., Rafighi, P. & Bagheri, R. Water oxidation by a soluble iron (III)–cyclen complex: new findings. Dalton Trans. 45, 2618–2623 (2016).
Annunziata, A. et al. Iron (III) complexes with cross‐bridged cyclams: synthesis and use in alcohol and water oxidation catalysis. Eur. J. Inorg. Chem. 2018, 3304–3311 (2018).
Das, B., Orthaber, A., Ott, S. & Thapper, A. Iron pentapyridyl complexes as molecular water oxidation catalysts: strong influence of a chloride ligand and pH in altering the mechanism. ChemSusChem 9, 1178–1186 (2016).
Kottrup, K. G. & Hetterscheid, D. G. Evaluation of iron-based electrocatalysts for water oxidation–an on-line mass spectrometry approach. Chem. Commun. 52, 2643–2646 (2016).
Wang, Z. Q., Wang, Z. C., Zhan, S. & Ye, J. S. A water-soluble iron electrocatalyst for water oxidation with high TOF. Appl. Catal., A 490, 128–132 (2015).
Kottrup, K. G., D’Agostini, S., van Langevelde, P. H., Siegler, M. A. & Hetterscheid, D. G. Catalytic activity of an iron-based water oxidation catalyst: substrate effects of graphitic electrodes. ACS Catal. 8, 1052–1061 (2018).
Codolà, Z. et al. Design of iron coordination complexes as highly active homogenous water oxidation catalysts by deuteration of oxidation-sensitive sites. J. Am. Chem. Soc. 141, 323–333 (2018).
Liu, T., Zhang, B. & Sun, L. Iron-based molecular water oxidation catalysts: abundant, Cheap, and Promising. Chem. Asia. J. 14, 31–43 (2019).
McKenzie, K. J. & Marken, F. Direct electrochemistry of nanoparticulate Fe2O3 in aqueous solution and adsorbed onto tin-doped indium oxide. Pure Appl. Chem. 73, 1885–1894 (2001).
Deutscher, J. et al. Water oxidation reaction mediated by an octanuclear iron‐oxo cluster. Eur. J. Inorg. Chem. 2018, 4925–4929 (2018).
Good, W. & Purdon, W. A. B. The photo and thermal oxidation of water by ferric iron. Chem. Ind. (London) 49, 1594–1595 (1995).
Elizarova, G. L., Matvienko, L. G., Lozhkina, N. V., Parmon, V. N. & Zamaraev, K. I. Homogeneous catalysts for dioxygen evolution from water. Water oxidation by trisbipyridylruthenium (III) in the presence of cobalt, iron and copper complexes. React. Kinet. Catal. Lett. 16, 191–194 (1981).
Yu.V. Karyakin., I.I. Angelov Chistye Khimicheskie Veschestva (Pure Chemical Reactants). Мoscow: Khimiya, (1974).
Parmon, V. N., Elizarova, G. L. & Kim, T. V. Spinels as heterogeneous catalysts for oxidation of water to dioxygen by tris-bipyridyl complexes of iron (III) and ruthenium (III). React. Kinet. Catal. Lett. 21, 195–197 (1982).
Kim, T. V., Elizarova, G. L. & Parmon, V. N. Catalytic oxidation of water to dioxygen by Ru(bpy)3+ in the presence of mixed iron and cobalt hydroxides. React. Kinet. Catal. Lett. 26, 57–60 (1984).
Najafpour, M. M., Moghaddam, A. N., Sedigh, D. J. & Hołyńska, M. A dinuclear iron complex with a single oxo bridge as an efficient water-oxidizing catalyst in the presence of cerium (IV) ammonium nitrate: new findings and current controversies. Catal. Sci. Technol. 4, 30–33 (2014).
Ren, X. et al. Homogeneous cobalt and iron oxide hollow nanocages derived from ZIF-67 etched by Fe species for enhanced water oxidation. Electrochim. Acta 296, 418–426 (2019).
Company, A. et al. Modeling the cis-oxo-labile binding site motif of non-heme iron oxygenases: water exchange and oxidation reactivity of a non-heme iron(IV)–oxo compound bearing a tripodal tetradentate ligand. Chem. Eur. J. 17, 1622–1634 (2011).
Kamiya, K., Kuwabara, A., Harada, T. & Nakanishi, S. Electrochemical formation of Fe (IV) = O derived from H2O2 on a hematite electrode as an active catalytic site for selective hydrocarbon oxidation reactions. ChemPhysChem. 20, 648–650 (2019).
Lefèvre, M., Proietti, E., Jaouen, F. & Dodelet, J.-P. Iron-based catalysts with improved oxygen reduction activity in polymer electrolyte fuel cells. Science 324, 71–74 (2009).
Smith, R. D. et al. Photochemical route for accessing amorphous metal oxide materials for water oxidation catalysis. Science 340, 60–63 (2013).
Smith, R. D., Prévot, M. S., Fagan, R. D., Trudel, S. & Berlinguette, C. P. Water oxidation catalysis: electrocatalytic response to metal stoichiometry in amorphous metal oxide films containing iron, cobalt, and nickel. J. Am. Chem. Soc. 135, 11580–11586 (2013).
Jang, J. W. et al. Enabling unassisted solar water splitting by iron oxide and silicon. Nat. Commun. 6, 7447 (2015).
Kim, J. Y. et al. Single-crystalline, wormlike hematite photoanodes for efficient solar water splitting. Sci. Rep. 3, 2681 (2013).
Subbaraman, R. et al. Trends in activity for the water electrolyser reactions on 3d M (Ni, Co, Fe, Mn) hydr (oxy) oxide catalysts. Nat. Mater. 11, 550–557 (2015).
Du, C. et al. Hematite‐based water splitting with low turn‐on voltages. Angew. Chem. Int. Ed. 52, 12692–12695 (2013).
Zhao, L. et al. Iron oxide embedded titania nanowires-An active and stable electrocatalyst for oxygen evolution in acidic media. Nano Energy 45, 118–126 (2018).
Okamura, M. et al. A pentanuclear iron catalyst designed for water oxidation. Nature 530, 465–468 (2016).
Bora, D. K., Braun, A. & Constable, E. C. “In rust we trust”. Hematite–the prospective inorganic backbone for artificial photosynthesis. Eng. Environ. Sci. 6, 407–425 (2013).
Jun, K., Lee, Y. S., Buonassisi, T. & Jacobson, J. M. High photocurrent in silicon photoanodes catalyzed by iron oxide thin films for water oxidation. Angew. Chem. Int. Ed. 51, 423–427 (2012).
Wu, Y. et al. Fast and simple preparation of iron‐based thin films as highly efficient water‐oxidation catalysts in neutral aqueous solution. Angew. Chem. Int. Ed. 54, 4870–4875 (2015).
Wilkins, R.G. Kinetics and mechanisms of reactions of transition metal complexes. VCH Publishers (1991).
Burke, M. S. et al. Revised oxygen evolution reaction activity trends for first-row transition-metal (oxy) hydroxides in alkaline media. J. Phys. Chem. Lett. 6, 3737–3742 (2015).
Ellis, W. C., McDaniel, N. D., Bernhard, S. & Collins, T. J. Fast water oxidation using iron. J. Am. Chem. Soc. 132, 10990–10991 (2010).
Carmona, F. et al. Ferritin iron uptake and release in the presence of metals and metalloproteins: chemical implications in the brain. Coord. Chem. Rev. 257, 2752–2764 (2013).
Jian, N., Dowle, M., Horniblow, R. D., Tselepis, C. & Palmer, R. E. Morphology of the ferritin iron core by aberration corrected scanning transmission electron microscopy. Nanotechnol. 27, 46LT02 (2016).
Harrison, P. M. & Arosio, P. The ferritins: molecular properties, iron storage function and cellular regulation. Biochim. Biophys. Acta –Bioenergetics 1275, 161–203 (1996).
Crichton, R. R. & Declercq, J. P. X-ray structures of ferritins and related proteins. Biochim. Biophys. Acta-General Subjects 1800, 706–718 (2010).
Drits, V. A., Sakharov, B. A., Salyn, A. L. & Manceau, A. Structural model for ferrihydrite. Clay Miner. 28, 185–207 (1993).
Ge, R. & Sun, X. Iron acquisition and regulation systems in Streptococcus species. Metallomics 6, 996–1003 (2014).
Neilands, J. B. Siderophores: structure and function of microbial iron transport compounds. J. Biol. Chem. 270, 26723–26726 (1995).
Hammer, N. D. & Skaar, E. P. Molecular mechanisms of Staphylococcus aureus iron acquisition. Ann. Rev. Microbiol. 65, 129–147 (2011).
Muthukumar, H. et al. Effect of iron doped Zinc oxide nanoparticles coating in the anode on current generation in microbial electrochemical cells. Int. J. Hydrog. Energy 44, 2407–2416 (2019).
Najafpour, M. M. et al. An aluminum/cobalt/iron/nickel alloy as a precatalyst for water oxidation. Int. J. Hydrog. Energy 43, 2083–2090 (2018).
Wang, A. et al. Tuning the oxygen evolution reaction on a nickel-iron alloy via active straining. Nanoscale 11, 426–30 (2019).
Yan, K., Lu, Y. & Jin, W. Facile synthesis of mesoporous manganese–iron nanorod arrays efficient for water oxidation. ACS Sustain. Chem. Eng. 4, 5398–5403 (2016).
Qi, K., Xie, Y., Wang, R., Liu, S. Y. & Zhao, Z. Electroless plating Ni-P cocatalyst decorated g-C3N4 with enhanced photocatalytic water splitting for H2 generation. Applied Surface Science 466, 847–853 (2019).
Qi, K., Liu, S. Y., Chen, Y., Xia, B. & Li, G. D. A simple post-treatment with urea solution to enhance the photoelectric conversion efficiency for TiO2 dye-sensitized solar cells. Solar Energy Materials and Solar Cells 183, 193–199 (2018).
Qin, Q. et al. Electrocatalysts: Low loading of RhxP and RuP on N, P codoped carbon as two trifunctional electrocatalysts for the oxygen and hydrogen electrode reactions. Adv. Energy Mater. 8(29), 1870130 (2018).
Qin, Q. et al. A tannic acid–derived N‐, P‐codoped carbon‐supported iron‐based nanocomposite as an advanced trifunctional electrocatalyst for the overall water splitting cells and zinc–air batteries. Adv. Energy Mater. 9, 1803312 (2019).
Qin, Q., Chen, L., Wei, T. & Liu, X. MoS2/NiS yolk–shell microsphere‐based electrodes for overall water splitting and asymmetric supercapacitor. Small, 1803639 (2018).
Acknowledgements
This work has been partly supported by the Institute for Advanced Studies in Basic Sciences and the National Elite Foundation. The authors thank Payam Salimi for the preparation of Figure S1. The authors are grateful to the Institute for Advanced Studies in Basic Sciences and the National Elite Foundation for financial support.
Author information
Authors and Affiliations
Contributions
M.M.N. proposed the concept. Z.A. and R.B. performed the experiments. All authors analyzed data and wrote the paper.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Abdi, Z., Bagheri, R., Song, Z. et al. Water oxidation by Ferritin: A semi-natural electrode. Sci Rep 9, 11499 (2019). https://doi.org/10.1038/s41598-019-47661-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-47661-z
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
