
Abstract
Pseudosarcomatous myofibroblastic proliferation is a descriptive term that designates a group of clinically indolent genitourinary lesions that most commonly arise in the urinary bladder. Given that pseudosarcomatous myofibroblastic proliferation may show morphologic overlap with inflammatory myofibroblastic tumor, the relationship, if any, between the two entities has been unclear. Moreover, pseudosarcomatous myofibroblastic proliferations are known to be positive for ALK immunohistochemistry in a subset of cases, although an inconsistent association with ALK rearrangement (ranging from 0 to 60%) has been reported. The objectives of this study were to determine the frequency of ALK rearrangement and to identify fusion partners using fluorescence in situ hybridization (FISH) and targeted RNA sequencing studies in a contemporary series of 30 pseudosarcomatous myofibroblastic proliferations of the urinary bladder, as well as to investigate ROS1 status by immunohistochemistry. ALK immunohistochemistry was positive in 70% (21/30) of pseudosarcomatous myofibroblastic proliferations; ROS1 immunohistochemistry was consistently negative (0/28). ALK rearrangements were detected by FISH in 86% (18/21) of cases, correlating with ALK immunohistochemical positivity in all but 3 cases. Of eight cases confirmed to be ALK rearranged by FISH, targeted RNA-sequencing detected FN1–ALK fusions in seven (88%) cases, which involved exons 20–26 of FN1 (5′) and exon 18-19 of ALK (3′). In conclusion, ALK rearrangements are frequent in pseudosarcomatous myofibroblastic proliferations, typically involving exon 19, and FN1 appears to be a consistent fusion partner. Given the significant clinicopathologic differences between inflammatory myofibroblastic tumor and pseudosarcomatous myofibroblastic proliferation, our findings provide further support for classification of pseudosarcomatous myofibroblastic proliferation as a distinct clinicopathologic entity, and propose the alternate terminology “pseudosarcomatous myofibroblastic neoplasm of the genitourinary tract.”
Similar content being viewed by others

Introduction
The term pseudosarcomatous myofibroblastic proliferation designates a group of lesions of uncertain nosology that show myofibroblastic differentiation and typically arise in the genitourinary tract or pelvic organs, with bladder being the most common site. First described in 1980 [1] and subsequently reported using a variety of names (including pseudosarcomatous fibromyxoid tumor [2, 3], inflammatory pseudotumor [4], inflammatory pseudosarcoma [5], visceral fasciitis, and postoperative spindle cell nodule [6]), this group of tumors are currently classified as inflammatory myofibroblastic tumors in the 4th edition World Health Organization (WHO) Classification of Tumors of the Urinary System and Male Genital Organs [7]. Nonetheless, tumors that are designated pseudosarcomatous myofibroblastic proliferation have the typical morphologic appearance of spindled myofibroblasts arranged in a loose “tissue culture”-like pattern that can be reminiscent of nodular fasciitis [8, 9], though some closely resemble conventional inflammatory myofibroblastic tumor. Larger series have described the morphologic spectrum of pseudosarcomatous myofibroblastic proliferation to encompasses cases with prominent mitotic activity, necrosis and infiltrative growth, and may be worrisome for low-grade sarcomas. However, pseudosarcomatous myofibroblastic proliferations lack metastatic potential and invariably show indolent clinical behavior [8,9,10,11].
The relationship between pseudosarcomatous myofibroblastic proliferation and inflammatory myofibroblastic tumor has not been entirely clear, and the ideal terminology and classification of pseudosarcomatous myofibroblastic proliferation has been uncertain. Both entities share some histologic and immunophenotypic features, including frequent expression of ALK protein by immunohistochemistry, the basis for which some authors consider pseudosarcomatous myofibroblastic proliferation to be classified as inflammatory myofibroblastic tumor [12]. However, while both entities can show local recurrences, only inflammatory myofibroblastic tumor has risk for metastasis (albeit low). The widely variable reported frequencies of ALK rearrangement in pseudosarcomatous myofibroblastic proliferations, ranging from 0 to ~70%, also adds to this controversy [8, 9, 12]. Inflammatory myofibroblastic tumors harbor ALK rearrangements in up to 60% of cases (mostly in young patients), with a heterogeneous group of fusion partners including TPM3, TPM4, CLTC, ATIC, RANBP2, and CARS [13,14,15,16,17]. Thus far, no concerted effort has been made to identify fusion partners in ALK-rearranged pseudosarcomatous myofibroblastic proliferation or clinicopathologically similar presumed inflammatory myofibroblastic tumors of the genitourinary tract, with limited targeted RNA-seq results reported in only two cases [9, 10, 12]. Alternate ROS1 fusions characterize a small subset of inflammatory myofibroblastic tumors [18], however ROS1 rearrangement has not been identified in pseudosarcomatous myofibroblastic proliferations [10].
The objectives of this study were to determine the prevalence of ALK gene rearrangement using fluorescence in situ hybridization (FISH), to identify specific fusion genes by targeted RNA sequencing, and to screen for ROS1 rearrangements by ROS1 immunohistochemistry, in a large cohort of pseudosarcomatous myofibroblastic proliferations of the urinary bladder.
Materials and methods
This study was performed with the approval of the Institutional Review Board of Brigham and Women’s Hospital, Boston, MA.
Cohort selection and histopathologic evaluation
Thirty cases diagnosed as pseudosarcomatous myofibroblastic proliferation arising in the bladder that had available archival unstained formalin-fixed paraffin-embedded (FFPE) slides were retrieved from the consultation files of one of the authors (C.D.M.F.). All diagnoses were rendered at the time of initial consultation and were based on established criteria [8, 9]. Hematoxylin and eosin-stained slides and immunohistochemical studies performed at the time of initial diagnosis were reviewed. In our practice, pseudosarcomatous myofibroblastic proliferations are classified separately from inflammatory myofibroblastic tumors, the latter based on characteristic features including hypercellularity, compact or cellular fascicular architecture, and/or a prominent lymphoplasmacytic infiltrate.
Immunohistochemistry
Immunohistochemistry was performed on 4 µm FFPE sections using the following antibodies and conditions: ALK (clone 5A4; Leica Biosystems, Buffalo Grove, IL, USA; 1:100 dilution; citrate buffer), ROS1 (clone D4D6; Cell Signaling Technology, Danver, MA, USA; 1:250 dilution; EDTA pressure cook), SMA (clone 1A4; Sigma, St Louis, MO, USA; 1:20,000; no pretreatment), desmin (clone DE-U-10; Sigma; 1:5000 dilution; citrate buffer and pressure cook), pan-keratin (MNF-116; Dako, Carpinteria, CA, USA; 1:700 dilution; 10 min protease digestion), AE1/AE3 (AE1+AE3; Dako, 1:200; 10 min protease digestion), and CAM5.2 (CAM5.2; Dako; 1:50; 10 min protease digestion). The Novolink Polymer detection system (Leica Biosystems) was used for signal detection. Staining was performed using a Link48 automated platform (Agilent, Santa Clara, CA, USA). Appropriate positive and negative controls were used throughout. Immunohistochemistry was annotated semi-quantitatively for staining intensity and extent (negative, no staining; focal, <10% of lesional cells; multifocal, ≥10% and <50%; diffuse, ≥50%), and the distribution of ALK and ROS1 staining (e.g., cytoplasmic, membranous, and nuclear) was recorded.
Fluorescence in situ hybridization
FISH analysis for ALK rearrangement was performed on all pseudosarcomatous myofibroblastic proliferations that were positive for ALK immunohistochemistry. In addition to the cases in this study, four pseudosarcomatous myofibroblastic proliferations that were previously reported by our group as negative for ALK rearrangement (via FISH analysis on tissue scrolls at the time) were re-tested [8]. FISH was performed on 4 µm FFPE tissue sections using LSI ALK dual color break-apart probes (Abbott Molecular, Abbott Park, IL) specific for the 5′ (red) and 3′ (green) regions of ALK at 2p23. Probe labeling and hybridization were performed following manufacturer’s directions (Abbott Molecular/Vysis, Inc.) according to standard protocols in our laboratory. Fifty individual, nonoverlapping interphase tumor nuclei with distinct nuclear boundaries were scored manually by a cytogeneticist (P.D.C.). Signals separated by one or more probe lengths were considered split signals, and cases showing split signals in more than 2% (>1/50) of tumor nuclei were considered positive for ALK rearrangement.
RNA sequencing
For ten cases, 4 µm sections of FFPE tumor were manually dissected from 2 to 5 glass slides per case for RNA extraction. RNA extraction and preparation were done using the ExpressArt FFPE Clear RNA Ready kit (Amsbio, Cambridge, MA), and total RNA was assessed using the Agilent 2100 Bioanalyzer and RNA 6000 Nano Kit.). RNA-Seq libraries were prepared with the TruSight RNA Fusion Panel (Illumina, San Diego, CA) with an input of 20–100 ng RNA per case. Each sample was sequenced with 76 base-pair paired-end reads on an Illumina MiSeq at 8 samples per flow cell (~3 million reads per sample). Results were analyzed using two pipelines; STAR aligner with Manta fusion caller through the Illumina Local Run Manager (v.1.3.0), and BOWTIE2 alignment with the JAFFA fusion caller [19, 20].
Results
Clinicopathologic features of the cohort
Patients in the cohort showed a wide age range (12–81 years), with median age of 47 years. There was a 2:1 male predominance (20 men, 10 women). All cases arose in the urinary bladder. Submitted clinical data available for 11 patients indicate 8 patients had a prior history of instrumentation or surgery, 3 of whom had urothelial carcinoma and 1 had prostatic adenocarcinoma. Seventeen [17] patients were symptomatic at presentation (most [13] reporting hematuria), and tumors were incidental findings in four patients. Two patients had a reported history of neurofibromatosis.
Microscopically, pseudosarcomatous myofibroblastic proliferations were characterized by myofibroblasts arranged in a “tissue culture-like” pattern, having palely eosinophilic cytoplasm with delicate processes varying from short to long and slender (Fig. 1a). Nuclei were elongated, ovoid or round, with an overall bland appearance, containing delicate chromatin and small to inconspicuous nucleoli (Fig. 1b). Tumor cells occasionally resembled strap-like rhabdomyoblasts. Two-thirds (20/30; 67%) of the lesions demonstrated an infiltrative growth pattern with extension through the muscularis propria (Fig. 1c). In one case, the tumor extended into perivesicular soft tissue and partially encased the seminal vesicle. Areas with increased cellularity and more epithelioid morphology were focally present in 4/30 (13.3%) cases (Fig. 2). The stroma was characteristically myxoid, with variable inflammatory infiltrates comprised of neutrophils, eosinophils, or lymphocytes. The degree of inflammation varied between tumors, from minimal (scattered inflammatory cells interspersed between tumor cells) in 13/30 (43.3%) cases to marked, to densely confluent infiltrates in 17/30 (56.7%) cases. While most infiltrates had a population of lymphocytes, numerous neutrophils and/or eosinophils were present in 24/30 cases (80%) and mast cells in 3 cases (10%); 3 (10%) cases had a lymphoplasmacytic infiltrate, though with fewer plasma cells than expected in inflammatory myofibroblastic tumor. Necrosis was seen in 18 cases, frequently in association with areas of surface ulceration. Overall, mitotic activity was low, with a median count of 4 mitotic figures per 10 high-power fields. Some cases showed more prominent mitotic activity, and 3 pseudosarcomatous myofibroblastic proliferations showed mitotic counts >19 per 10 high-power fields. In such cases, these more mitotically active areas were often focal and associated with necrosis and/or inflammation. Although some pseudosarcomatous myofibroblastic proliferations showed scattered foci of myofibroblasts having enlarged nuclei and more prominent nucleoli, other features suggestive of malignancy such as frank pleomorphism, nuclear hyperchromasia, and atypical mitotic figures were not identified.

Tumors typically show slender myofibroblasts arranged in a “tissue culture-like” pattern; the stroma is typically myxoid and there is usually a mixed inflammatory infiltrate with neutrophils, eosinophils, or lymphocytes, and only rare (if any) plasma cells (a). Tumor cells have palely eosinophilic cytoplasm with long tapering processes, elongated nuclei with delicate chromatin and inconspicuous or small round nucleoli. Mitotic activity is present in most cases (b). Lesions usually involve the muscularis propria (c).

Some examples show foci of increased cellularity (a), where mitotic activity can be higher. A subset of tumors may also show epithelioid cytomorphology with enlarged round nuclei, prominent nucleoli, and abundant cytoplasm in hypercellular areas (b).
The overall immunohistochemical features are summarized in Table 1. Pseudosarcomatous myofibroblastic proliferations were invariably positive for desmin (30/30) and frequently positive for smooth muscle actin (28/29; 96.6%). Overall, keratin (pan-keratin, AE1/AE3, and/or CAM5.2) expression was seen in ~67% (18/27) of cases, most often in a multifocal distribution. ALK protein was positive in 21/30 (70%) of the cases, most being diffuse (14/21; 70%) or multifocal (5/21; 25%) in extent. All ALK-positive pseudosarcomatous myofibroblastic proliferations showed either cytoplasmic and membranous (11/21; 55%) or cytoplasmic (9/21; 55%) staining for ALK immunohistochemistry (Fig. 3). ROS1 immunohistochemistry was consistently negative in all lesions tested (0/28).

A significant proportion (70%) of pseudosarcomatous myofibroblastic proliferations show cytoplasmic and membranous (a) or cytoplasmic (b) positivity for ALK immunohistochemistry.
ALK fluorescence in situ hybridization and RNA sequencing
Table 2 summarizes the immunohistochemical, FISH, and RNA-Seq results for the cohort. All pseudosarcomatous myofibroblastic proliferations positive for ALK immunohistochemistry underwent FISH analysis. In total, ALK rearrangement was detected in 18/21 (86%) cases (Fig. 4a). Given the high frequency of ALK rearrangements in our current cohort, we retested four additional pseudosarcomatous myofibroblastic proliferations that were previously reported by our group as negative for ALK rearrangement [8], and repeat FISH analysis revealed the presence of ALK rearrangement in 1/4 (25%) of these cases.

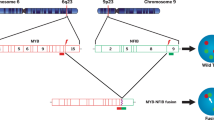
FISH break-apart probes showing split signals for ALK (red, 5′; green, 3′) (a) confirmed the presence of ALK rearrangement in pseudosarcomatous myofibroblastic proliferations, correlating with ALK immunohistochemistry expression in 86% of cases. RNA-Seq identified the FN1–ALK fusions in 7/8 cases tested; a schematic illustrating exon a nd protein structure of ALK (top), FN1 (middle), and the FN1–ALK fusion product, with the predicted breakpoints are indicated by black arrowheads (b). All fusions involved exon 19 of ALK, and the chimeric protein includes the ALK kinase domain and transmembrane portion of the protein.
Ten pseudosarcomatous myofibroblastic proliferations underwent RNA sequencing, including eight cases positive for both ALK protein expression and ALK rearrangement by FISH, one case positive for ALK protein expression but negative for ALK rearrangement by FISH, and one case negative for ALK by immunohistochemistry. RNA sequencing failed due to poor quality RNA on the latter two cases, which were the oldest cases tested (from 2014).
Seven of the 8 remaining cases (87.5%) showed FN1–ALK fusions encompassing ALK exons 19–29 (in 6/7 cases) and 18–29 (in 1 case) with predicted breakpoints in introns 18 and 17, respectively (Fig. 4b). The predicted breakpoints of FN1 were more variable and involved introns 20, 22, 23, 24, and 26, rendering fusion products that encompass exons 1–20 in 3 cases and exons 1–22, 1–23, 1–24, and 1–26 in one case each. All FN1–ALK fusions were in-frame, showing a concordant orientation of the fusion partners with 5′ FN1 and a 3′ ALK sequences. Importantly, all these fusion products include the intracytoplasmic C-terminal domain of ALK (exons 20–29), which contains the kinase domain, as well as the transmembrane portion of the protein (exon 19) [21, 22]. No fusion was detected using automated fusion callers in the eighth pseudosarcomatous myofibroblastic proliferation by RNAseq, although it was positive for ALK immunohistochemistry and ALK-rearranged by FISH.
Discussion
The term pseudosarcomatous myofibroblastic proliferation was first introduced in 1980 in a case report to describe what was thought to be a reactive myofibroblastic proliferation with pseudosarcomatous morphologic features arising in the urinary bladder [1]. Over the next two decades, similar lesions were reported under a variety of different designations, including pseudosarcomatous fibromyxoid tumor [2, 3], inflammatory pseudotumor [4], inflammatory pseudosarcoma [5], and inflammatory postoperative spindle cell nodule [6]; notably, there does not appear to be any clinicopathologic distinction between tumors with and without prior instrumentation in this group [9]. The designation “pseudosarcomatous” refers to the propensity of these lesions to show extension through bladder wall, prominent mitotic activity and, in context, morphologic mimicry of a myogenic sarcoma. Keratins, desmin, and smooth muscle actin are frequently positive. Based on their shared morphologic and clinical features, these lesions are now collectively classified as pseudosarcomatous myofibroblastic proliferations.
The more confusing issue involves the distinction between pseudosarcomatous myofibroblastic proliferation and inflammatory myofibroblastic tumor. Some authors have considered pseudosarcomatous myofibroblastic proliferations to be related to (or synonymous with) inflammatory myofibroblastic tumor, given the occasional shared morphologic features and that a significant proportion of both entities show expression of ALK protein. Adding to the confusion for the classification of pseudosarcomatous myofibroblastic proliferation is the inconsistent association with ALK rearrangement reported in the literature [8, 9, 23]. Currently, pseudosarcomatous myofibroblastic proliferations are classified as inflammatory myofibroblastic tumors in the 4th edition WHO Classification of Tumors of the Urinary System and Male Genital Organs [7]. However, we and other authors have considered pseudosarcomatous myofibroblastic proliferations and classic inflammatory myofibroblastic tumors to represent distinct entities based on their differences in their clinical and pathologic features. Inflammatory myofibroblastic tumor more often affects children and young adults, while pseudosarcomatous myofibroblastic proliferation arises most commonly during adulthood and shows a clear male predominance. Most pseudosarcomatous myofibroblastic proliferations show “tissue culture-like” architecture and are relatively hypocellular, compared to the more hypercellular fascicular arrangement of tumor cells in inflammatory myofibroblastic tumor. In addition, the lesional cells of pseudosarcomatous myofibroblastic proliferation tend to be longer with more delicate tapering cytoplasmic processes. Pseudosarcomatous myofibroblastic proliferations also have prominent myxoid stroma, and plasma cells are rare within the mixed inflammatory infiltrates. From a clinical perspective, tumors classified as pseudosarcomatous myofibroblastic proliferations invariably follow an indolent clinical course, showing nondestructive local recurrences in up to 20% of cases and virtually no metastatic potential [8,9,10,11]. Overall, inflammatory myofibroblastic tumor can also show local recurrence in up to a third of cases, however up to 5% of inflammatory myofibroblastic tumors (including those in abdominal/pelvis sites) can show an aggressive clinical course and metastasize [24]. Inflammatory myofibroblastic tumors in the bladder are uncommon; among ~400 inflammatory myofibroblastic tumors in a large soft tissue pathology consultation archive of one of the authors (C.D.M.F.), 6 arose in the bladder (5 of which were in pediatric patients).
The correlation between ALK protein expression and ALK rearrangement in pseudosarcomatous myofibroblastic proliferations has been unclear, and is further complicated by the fact that some studies of bladder inflammatory myofibroblastic tumor have included cases that may be better classified as pseudosarcomatous myofibroblastic proliferation. The use of different immunohistochemical antibody clones and conditions can also affect results. In 2006, our group reported a series of pseudosarcomatous myofibroblastic proliferations, of which six cases showed ALK protein expression but lacked ALK rearrangement by FISH analysis [8]. A subsequent study by Harik et al. identified ALK rearrangements in 4/6 (67%) of pseudosarcomatous myofibroblastic proliferations [9], followed by Montgomery et al. who reported an overall frequency of 72% (13/18) in a series of bladder inflammatory myofibroblastic tumors that included pseudosarcomatous myofibroblastic proliferations [12]. In this current series, we found ALK protein expression in 70% of pseudosarcomatous myofibroblastic proliferations, and among immunohistochemistry-positive cases 86% (18/21) had ALK rearrangements. Our current findings prompted us to re-test four ALK immunohistochemistry-positive/FISH-negative cases from our prior 2006 study, and ALK rearrangement was identified in one (1/4, 25%) case. These discordant FISH results are attributed to subsequent changes in sample preparation in our laboratory. Previously, 50 intact nuclei were isolated from 50 µm scrolls, which comprised a mixture of normal and tumor cells, and distinguishing whether the scored nuclei were from tumor cells or non-lesional cells was not possible after probe hybridization. Currently, FISH analysis is performed on 4 µm tissue section slides on which nuclei are scored only in areas of tumor designated by a pathologist on the corresponding hematoxylin and eosin-stained slide. It is still unclear why FISH analysis was negative for ALK rearrangement in rare pseudosarcomatous myofibroblastic proliferations that are positive for ALK immunohistochemistry. One possibility is that some cases harbor cryptic structural variants that cannot be resolved by FISH. Unfortunately, attempted RNA sequencing on one of these ALK immunohistochemistry-positive/FISH-negative cases in our cohort was unsuccessful, likely due to the age of the sample. A small but significant fraction of ALK-negative inflammatory myofibroblastic tumors have ROS1 rearrangements which can be detected by ROS1 immunohistochemistry with high sensitivity and specificity [25, 26]. One recent study demonstrated that ROS1 immunohistochemistry is negative in a cohort of 8 ALK-negative pseudosarcomatous myofibroblastic proliferations [10]; similarly in this present study, all 28 cases tested were negative for ROS1.
RNA sequencing revealed that the FN1 gene was a consistent fusion partner to ALK in pseudosarcomatous myofibroblastic proliferations. FN1 is located at 2q35 and encodes fibronectin, a major extracellular matrix protein that is ubiquitously expressed in a wide variety of tissues. Interestingly, experimental models suggest a reciprocal relationship between fibronectin production and the acquisition and maintenance of a myofibroblastic phenotype. In vitro, the presence of fibronectin is required for TGFβ-induced myofibroblatic differentiation [27, 28]. In this setting, fibronectin induces myofibroblastic differentiation by binding to integrin receptors and activating focal adhesion kinases [29, 30]. In turn, myofibroblasts express, secrete and assemble fibronectin in the extracellular matrix [31, 32]. Similar to other ALK fusion genes, the mechanism of ALK activation may likely be due to FN1 providing strong promoter function and facilitating dimerization of ALK receptors [33]. Interestingly, these FN1–ALK fusions include both the transmembrane and cytoplasmic domains of the ALK protein, and we observed ALK staining in pseudosarcomatous myofibroblastic proliferations to be localized to the cytoplasm, with 11 (55%) showing membranous staining as well. FN1 has been identified as a fusion partner to ALK in rare cases of genitourinary inflammatory myofibroblastic tumor, and only in a single case among inflammatory myofibroblastic tumors at other locations. These reported genitourinary cases include three bladder tumors diagnosed as inflammatory myofibroblastic tumor (two arising in 8-year-old and 26-year-old female patients [25] and one in a 12-year-old male patient [34]) and two uterine inflammatory myofibroblastic tumors in a 50 years old and 39 years old, the latter arising during pregnancy [35]. Notably, these reported cases also harbor identical FN1–ALK fusion types with involvement of ALK exon 19. While these cases are intriguing, further information is not available to determine whether some (if not all) reported cases should have been classified as pseudosarcomatous myofibroblastic proliferations. FN1–ALK fusions have also been reported in gastrointestinal leiomyomas and a so-called “stromal-sarcoma” of the ovary [35,36,37], involving FN1 exons 1–41/42 and ALK exons 16–29 in the former, and FN1 exons 1–23 and ALK exons 19–29 in the latter. Recently, in a series of pediatric inflammatory myofibroblastic tumors, FN1–ROS1 fusion was reported in a single case of inflammatory myofibroblastic tumor arising in the lung of a 2-year-old male [38].
The consistency of fusions involving ALK exon 19 and FN1 exons 20–26 identified in pseudosarcomatous myofibroblastic proliferation is quite striking, and further supports that ALK-positive pseudosarcomatous myofibroblastic proliferations of the urinary bladder represent a distinct entity from conventional inflammatory myofibroblastic tumor. Inflammatory myofibroblastic tumor harbors a wide range of ALK fusion genes and variants [18, 25]. In the latter, the most common fusions are TPM3-ALK and CLTC-ALK, and small subsets harbor alternative ALK fusion partners and ROS1 and PDGFRB rearrangements [18, 25]. Furthermore, most inflammatory myofibroblastic tumors with ALK rearrangement involve exon 20, while all pseudosarcomatous myofibroblastic proliferations sequenced in our series showed ALK fusions involving exon 18 or 19 which has only been rarely seen in inflammatory myofibroblastic tumors, many in genitourinary sites [25, 34, 35]. Interestingly, ALK fusions involving exon 19 were found in 9/11 uterine inflammatory myofibroblastic tumors published by Haimes et al. [35], two of which had FN1–ALK. Nearly all patients had been treated by hysterectomy and were reported to be disease-free. Although our findings suggest that FN1–ALK fusion may be a defining feature of pseudosarcomatous myofibroblastic proliferation, it is likely not exclusive, and it should be emphasized that the diagnosis of pseudosarcomatous myofibroblastic proliferation versus inflammatory myofibroblastic tumor should rely first on clinical and pathologic features, rather than on the basis of gene fusion present. Table 3 summarizes the distinctive clinicopathologic features of pseudosarcomatous myofibroblastic proliferation and inflammatory myofibroblastic tumor.
One limitation of our study is the lack of molecular data for the ALK immunohistochemistry-negative cases. We attempted to perform RNA sequencing in a pseudosarcomatous myofibroblastic proliferation that was negative for ALK expression and in one that was positive for ALK protein expression but negative for ALK rearrangement, however both samples were older than 5 years and sequencing was unsuccessful due to RNA degradation. Because all remaining ALK-negative samples were even older, we desisted from pursuing molecular testing of these cases. Further studies are needed to determine if ALK-negative pseudosarcomatous myofibroblastic proliferations harbor other characteristic genetic alterations.
The presence of consistent FN1–ALK fusions in pseudosarcomatous myofibroblastic proliferations supports their classification as a true neoplasm and, in the context of ALK exon 19 rearrangement, suggests that these are distinct from conventional inflammatory myofibroblastic tumors. Given the differences in histopathologic features and clinical behavior, we believe that including pseudosarcomatous myofibroblastic proliferations under the spectrum of inflammatory myofibroblastic tumor is not ideal. Classifying these lesions as inflammatory myofibroblastic tumor may lead clinicians to assume a small but not insignificant risk of metastases (~5%), thus recognizing these tumors as a distinct entity would avoid overtreatment. Revised diagnostic terminology that better reflects the neoplastic (but benign) nature of pseudosarcomatous myofibroblastic proliferation should be considered. We propose “pseudosarcomatous myofibroblastic neoplasm of the genitourinary tract,” hoping that this new designation will help clarify prior uncertainties and controversies regarding the relationship between these lesions and inflammatory myofibroblastic tumors.
References
Roth JA. Reactive pseudosarcomatous response in urinary bladder. Urology. 1980;16:635–7.
Ro JY, el-Naggar AK, Amin MB, Sahin AA, Ordonez NG, Ayala AG. Pseudosarcomatous fibromyxoid tumor of the urinary bladder and prostate: immunohistochemical, ultrastructural, and DNA flow cytometric analyses of nine cases. Hum Pathol. 1993;24:1203–10.
Ro JY, Ayala AG, Ordóñez NG, Swanson DA, Babaian RJ. Pseudosarcomatous fibromyxoid tumor of the urinary bladder. Am J Clin Pathol. 1986;86:583–90.
Nochomovitz LE, Orenstein JM. Inflammatory pseudotumor of the urinary bladder-possible relationship to nodular fasciitis. Two case reports, cytologic observations, and ultrastructural observations. Am J Surg Pathol. 1985;9:366–73.
Gugliada K, Nardi PM, Borenstein MS, Torno RB. Inflammatory pseudosarcoma (pseudotumor) of the bladder. Radiology. 1991;179:66–8.
Proppe KH, Scully RE, Rosai J. Postoperative spindle cell nodules of genitourinary tract resembling sarcomas. A report of eight cases. Am J Surg Pathol. 1984;8:101–8.
Moch HHP, Ulbright TM, Reuter VE, (eds). World Health Organization Classification of tumours of the urinary system and male genital organs. 4th ed. Lyon: IARC; 2016.
Hirsch MS, Dal Cin P, Fletcher CD. ALK expression in pseudosarcomatous myofibroblastic proliferations of the genitourinary tract. Histopathology. 2006;48:569–78.
Harik LR, Merino C, Coindre JM, Amin MB, Pedeutour F, Weiss SW. Pseudosarcomatous myofibroblastic proliferations of the bladder: a clinicopathologic study of 42 cases. Am J Surgical Pathol. 2006;30:787–94.
Jebastin JAS, Smith SC, Perry KD, Gupta NS, Alanee S, Carskadon S, et al. Pseudosarcomatous myofibroblastic proliferations of the genitourinary tract are genetically different from nodular fasciitis and lack USP6, ROS1 and ETV6 gene rearrangements. Histopathology. 2018;73:321–6.
Albores-Saavedra J, Manivel JC, Essenfeld H, Dehner LP, Drut R, Gould E, et al. Pseudosarcomatous myofibroblastic proliferations in the urinary bladder of children. Cancer. 1990;66:1234–41.
Montgomery EA, Shuster DD, Burkart AL, Esteban JM, Sgrignoli A, Elwood L, et al. Inflammatory myofibroblastic tumors of the urinary tract: a clinicopathologic study of 46 cases, including a malignant example inflammatory fibrosarcoma and a subset associated with high-grade urothelial carcinoma. Am J Surg Pathol. 2006;30:1502–12.
Lawrence B, Perez-Atayde A, Hibbard MK, Rubin BP, Dal Cin P, Pinkus JL, et al. TPM3-ALK and TPM4-ALK oncogenes in inflammatory myofibroblastic tumors. Am J Pathol. 2000;157:377–84.
Bridge JA, Kanamori M, Ma Z, Pickering D, Hill DA, Lydiatt W, et al. Fusion of the ALK gene to the clathrin heavy chain gene, CLTC, in inflammatory myofibroblastic tumor. Am J Pathol. 2001;159:411–5.
Debelenko LV, Arthur DC, Pack SD, Helman LJ, Schrump DS, Tsokos M. Identification of CARS-ALK fusion in primary and metastatic lesions of an inflammatory myofibroblastic tumor. Lab Investig. 2003;83:1255–65.
Debiec-Rychter M, Marynen P, Hagemeijer A, Pauwels P. ALK-ATIC fusion in urinary bladder inflammatory myofibroblastic tumor. Genes Chromosom Cancer. 2003;38:187–90.
Patel AS, Murphy KM, Hawkins AL, Cohen JS, Long PP, Perlman EJ, et al. RANBP2 and CLTC are involved in ALK rearrangements in inflammatory myofibroblastic tumors. Cancer Genet Cytogenet. 2007;176:107–14.
Antonescu CR, Suurmeijer AJ, Zhang L, Sung YS, Jungbluth AA, Travis WD, et al. Molecular characterization of inflammatory myofibroblastic tumors with frequent ALK and ROS1 gene fusions and rare novel RET rearrangement. Am J Surg Pathol. 2015;39:957–67.
Liu S, Tsai WH, Ding Y, Chen R, Fang Z, Huo Z, et al. Comprehensive evaluation of fusion transcript detection algorithms and a meta-caller to combine top performing methods in paired-end RNA-seq data. Nucleic Acids Res. 2016;44:e47.
Lee JH, Huang HY, Lan J, Hwang CC, Liu CY. Cutaneous syncytial myoepithelioma: a case report with emphasis on the differential diagnosis of problematic dermal tumors. Oncol Lett. 2015;9:2275–7.
Flicek P, Ahmed I, Amode MR, Barrell D, Beal K, Brent S, et al. Ensembl 2013. Nucleic Acids Res. 2013;41(Database issue):D48–55.
Le AT, Varella-Garcia M, Doebele RC. Oncogenic fusions involving exon 19 of ALK. J Thorac Oncol. 2012;7:e44.
Tsuzuki T, Magi-Galluzzi C, Epstein JI. ALK-1 expression in inflammatory myofibroblastic tumor of the urinary bladder. Am J Surg Pathol. 2004;28:1609–14.
Coffin CM, Hornick JL, Fletcher CD. Inflammatory myofibroblastic tumor: comparison of clinicopathologic, histologic, and immunohistochemical features including ALK expression in atypical and aggressive cases. Am J Surg Pathol. 2007;31:509–20.
Lovly CM, Gupta A, Lipson D, Otto G, Brennan T, Chung CT, et al. Inflammatory myofibroblastic tumors harbor multiple potentially actionable kinase fusions. Cancer Discov. 2014;4:889–95.
Hornick JL, Sholl LM, Dal Cin P, Childress MA, Lovly CM. Expression of ROS1 predicts ROS1 gene rearrangement in inflammatory myofibroblastic tumors. Mod Pathol. 2015;28:732–9.
Lygoe KA, Wall I, Stephens P, Lewis MP. Role of vitronectin and fibronectin receptors in oral mucosal and dermal myofibroblast differentiation. Biol Cell. 2007;99:601–14.
Serini G, Bochaton-Piallat ML, Ropraz P, Geinoz A, Borsi L, Zardi L, et al. The fibronectin domain ED-A is crucial for myofibroblastic phenotype induction by transforming growth factor-beta1. J Cell Biol. 1998;142:873–81.
Greenberg RS, Bernstein AM, Benezra M, Gelman IH, Taliana L, Masur SK. FAK-dependent regulation of myofibroblast differentiation. FASEB J. 2006;20:1006–8.
Thannickal VJ, Lee DY, White ES, Cui Z, Larios JM, Chacon R, et al. Myofibroblast differentiation by transforming growth factor-beta1 is dependent on cell adhesion and integrin signaling via focal adhesion kinase. J Biol Chem. 2003;278:12384–9.
Torr EE, Ngam CR, Bernau K, Tomasini-Johansson B, Acton B, Sandbo N. Myofibroblasts exhibit enhanced fibronectin assembly that is intrinsic to their contractile phenotype. J Biol Chem. 2015;290:6951–61.
Ignotz RA, Massagué J. Transforming growth factor-beta stimulates the expression of fibronectin and collagen and their incorporation into the extracellular matrix. J Biol Chem. 1986;261:4337–45.
Mano H. ALKoma: a cancer subtype with a shared target. Cancer Discov. 2012;2:495–502.
Ouchi K, Miyachi M, Tsuma Y, Tsuchiya K, Iehara T, Konishi E, et al. FN1: a novel fusion partner of ALK in an inflammatory myofibroblastic tumor. Pediatr Blood Cancer. 2015;62:909–11.
Haimes JD, Stewart CJR, Kudlow BA, Culver BP, Meng B, Koay E, et al. Uterine inflammatory myofibroblastic tumors frequently harbor ALK fusions With IGFBP5 and THBS1. Am J Surg Pathol. 2017;41:773–80.
Panagopoulos I, Gorunova L, Lund-Iversen M, Lobmaier I, Bjerkehagen B, Heim S. Recurrent fusion of the genes FN1 and ALK in gastrointestinal leiomyomas. Mod Pathol. 2016;29:1415–23.
Ren H, Tan ZP, Zhu X, Crosby K, Haack H, Ren JM, et al. Identification of anaplastic lymphoma kinase as a potential therapeutic target in ovarian cancer. Cancer Res. 2012;72:3312–23.
Lopez-Nunez O, John I, Panasiti RN, Ranganathan S, Santoro L, Grélaud D, et al. Infantile inflammatory myofibroblastic tumors: clinicopathological and molecular characterization of 12 cases. Mod Pathol. 2020;33:576–90.
Funding
This work was funded by departmental resources.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Acosta, A.M., Demicco, E.G., Dal Cin, P. et al. Pseudosarcomatous myofibroblastic proliferations of the urinary bladder are neoplasms characterized by recurrent FN1–ALK fusions. Mod Pathol 34, 469–477 (2021). https://doi.org/10.1038/s41379-020-00670-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-020-00670-0
