
Abstract
Androgen receptor (AR), a ligand-activated transcription factor, is a master regulator in the development and progress of prostate cancer (PCa). A major challenge for the clinically used AR antagonists is the rapid emergence of resistance induced by the mutations at AR ligand binding domain (LBD), and therefore the discovery of novel anti-AR therapeutics that can combat mutation-induced resistance is quite demanding. Therein, blocking the interaction between AR and DNA represents an innovative strategy. However, the hits confirmed targeting on it so far are all structurally based on a sole chemical scaffold. In this study, an integrated docking-based virtual screening (VS) strategy based on the crystal structure of the DNA binding domain (DBD) of AR was conducted to search for novel AR antagonists with new scaffolds and 2-(2-butyl-1,3-dioxoisoindoline-5-carboxamido)−4,5-dimethoxybenzoicacid (Cpd39) was identified as a potential hit, which was competent to block the binding of AR DBD to DNA and showed decent potency against AR transcriptional activity. Furthermore, Cpd39 was safe and capable of effectively inhibiting the proliferation of PCa cell lines (i.e., LNCaP, PC3, DU145, and 22RV1) and reducing the expression of the genes regulated by not only the full-length AR but also the splice variant AR-V7. The novel AR DBD-ARE blocker Cpd39 could serve as a starting point for the development of new therapeutics for castration-resistant PCa.
Similar content being viewed by others


Introduction
Prostate cancer (PCa) is one of the major fatal cancers for males worldwide. Androgen deprivation therapy along with the use of potent anti-androgens has been the cornerstone of treatment for PCa [1]. However, it is inevitable that PCa would eventually develop into castration resistant PCa (CRPC) due to the restoration of androgen receptor (AR) signaling [2,3,4]. Considering that AR signaling is crucial for PCa at all stages, AR antagonists are recognized as one of the most effective ways to treat PCa [5, 6]. Like other nuclear receptors, such as glucocorticoid receptor (GR), estrogen receptor (ER), progesterone receptor (PR), and mineralocorticoid receptor (MR), AR consists of four functional domains, including the N-terminal transcriptional activation domain (NTD), the conserved DNA binding domain (DBD), the hinge region, and the C-terminal ligand binding domain (LBD) [6, 7]. To date, all of the approved AR antagonists target the ligand binding pocket (LBP) of the LBD. Among them, flutamide (PubChem CID: 3397), bicalutamide (PubChem CID: 2375) and enzalutamide (PubChem CID: 15951529) have been widely used for the treatment of androgen-dependent PCa [8, 9]. Recently, another two AR antagonists apalutamide (ARN-509, an enzalutamide derivative) (PubChem CID: 24872560) and darolutamide (ODM-201) (PubChem CID: 67171867) were approved for the treatment of non-metastatic CRPC [9, 10]. However, a broad spectrum of studies demonstrated that the potency of conventional AR antagonists would be suffered from the rapid emergence of drug resistance [11]. A number of mutations at the AR LBP would induce resistance to first-generation antiandrogens, such as bicalutamide and hydroxyflutamide (PubChem CID: 91649) [11, 12]. A phase III study showed that the F876L mutation in the LBP of AR could induce resistance to enzalutamide and apalutamide [13]. In addition, the overexpression of AR splices variants (ARVs) without a part of or even the entire LBD in PCa specimens was observed [11, 14]. The predominant ARVs detected in CPRC patients include AR-V3, AR-V7 and AR-V9, and their expression levels are significantly high in 22RV1 and CWR-R1 CRPC cell lines [15], suggesting that ARVs would most likely play a critical role in the development of CRPC. Some ARVs are also implicated in the development of drug resistance by engaging the AR chromatin-binding sites and driving the AR transcriptional program in a constitutive and ligand-independent manner [3, 11]. Therefore, the discovery of novel AR antagonists is still quite urgent.
In recent years, several other targeting sites on AR, such as the action function 2 (AF2) site and the binding function 3 (BF3) site on the LBD, and the DNA binding site on the DBD, have attracted increasing attentions [16,17,18]. Targeting these sites may help overcome mutation-induced resistance to traditional AR antagonists targeting the LBP. The AR DBD contains two zinc finger regions, which contributes to DNA binding and homodimerization (Fig. 1a). The P-box at the N terminus of the α-helix in the first zinc finger can insert directly into the DNA major groove of androgen response element (ARE). The second zinc finger with the D-box interacts with another DBD monomer with a head-to-head arrangement [19, 20]. It is deduced that targeting the DBD can disrupt either the formation of the AR dimer or the interaction between the DBD and ARE, or both, and then suppress AR transcriptional activity. Recently, Jones et al. found that VPC-14337 (pyrvinium) (PubChem CID: 54680693) could strongly inhibit the full-length AR and splice variants, and computational modeling was performed and predicted that VPC-14337 functioned through the DBD [21]. Li et al. conducted structure-based virtual screening (SBVS) and discovered several AR antagonists targeting the P-box of the DBD in micromole concentration range [22]. Then, through the structural optimization of the most active hit, they identified a better candidate, Vpc-14228 (PubChem CID: 727584), with eGFP IC50 and PSA IC50 of 0.33 µM and 0.28 µM, respectively (Fig. 1b). Further structure-activity relationship (SAR) analysis of the candidate led to the discovery of VPC-14449 (PubChem CID: 91754488) (eGFP IC50 = 0.12 ± 0.01 µM and PSA IC50 = 0.17 µM) [22]. The project resulted in the largest academic licensing deal in Canadian history, totaling $142 M [23]. Recently, they discovered a series of AR DBD dimer blockers that possibly target the D-box of the DBD, and the best hit compound VPC-17005 showed submicromolar AR antagonistic activity (eGFP IC50 = 0.734 µM and PSA IC50 = 0.691 µM) [24]. To develop a novel drug, the discovery of a new chemical scaffold is normally the initial and vital step, and then structural modification plays an important part to push it forwards. Although the efforts have been dedicated to the discovery of AR antagonists toward the AR DBD, the potential hits confirmed disrupting the binding of AR DBD to ARE are all structurally based on a sole scaffold and no candidate has been pushed into the clinical trials, therefore the discovery of AR DBD antagonist with novel scaffolds is required.

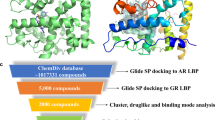
a The structure of the AR DBD. b The structures of the two bioactive compounds targeting the AR DBD and the structure of Cpd39. c Workflow of the integrated docking-based VS. d The AR transcriptional inhibition of Cpd39 in LNCaP-ARR2PB-eGFP (n = 4). e The PSA suppression by these compounds was evaluated by measuring the PSA secreted into the media using the same LNCaP-ARR2PB-eGFP cells (n = 4). f The 3D presentation of the interactions between Cpd39 and the AR DBD. Cpd39 is colored green, and the surrounding residues are colored purple. g The electrostatic potential of the binding site. The residues with the positive electrostatic potential are colored blue, and those with the negative electrostatic potential are colored red. h The 2D presentation of the interactions between Cpd39 and the AR DBD.
In this study, to discover new-scaffold antagonists targeting the AR DBD, an integrated docking-based virtual screening (VS) toward the DBD was conducted, and 61 compounds were selected for bioassays. Among the identified hits, 2-(2-butyl-1,3-dioxoisoindoline-5-carboxamido)−4,5-dimethoxybenzoic acid (Cpd39) exhibited the best potency, providing a new scaffold for the development of new therapeutics against CRPC.
Materials and methods
Protein preparation and binding site detection
The crystal structure of the AR DBD dimer bound to two hexameric half-site responds elements (PDB entry: 1R4I) [20] was used the template structure for VS. As the sequences of rat and human AR DBD are identical, the rat crystal structure was directly used as the human one just after the residues were renumbered and the mutated CYS552 was remutated. The Protein Preparation Wizard in Schrödinger 2017 was utilized to prepare the protein [25], including assigning bond orders, adding hydrogen atoms, filling in missing side chains, and minimizing the system with the OPLS2005 [26, 27] force field until the root-mean-square deviation (RMSD) of heavy atoms converged to 0.30 Å. The protonation states of residues at pH = 7.0 were determined by PROPKA [28], and only the single protomer was remained for the final VS. If the used docking program had its own protein preparation function, it would be employed to handle the protein further.
The potential binding site in the DBD was determined by the Site Finder module in Molecular Operating Environment (MOE) just as Li et al. did in their study [22]. The identified binding site located at the AR DBD-ARE binding interface was embraced by the residues Ser579, Val582, Phe583, Arg586, Arg609, Lys610, Pro613, and Arg616.
Docking-based virtual screening
Three molecular docking programs, namely Glide [29], GOLD [30], and AutoDock Vina [31], were used in docking-based VS. All the parameters were set to the default values, unless otherwise noted as followed.
Glide
The Receptor Grid Generation utility of Glide was used to generate the receptor grid, which is located in a binding box with the size of 10 Å × 10 Å × 10 Å centered on the centroid of the residues that form the pocket. The standard precision (SP) scoring mode was used in molecular docking.
GOLD
The protein was prepared by the built-in protein preparation function, and the binding site was defined by all the atoms within 10 Å of the pocket. The genetic algorithm (GA) method with the “automatic” settings and the default Piecewise Linear Potential (CHEMPLP) scoring function were utilized for sampling and scoring, respectively.
Autodock vina
The structures of the protein and ligands were converted into the pdbqt formats by AutoDockTools, along with the addition of hydrogen atoms, assignment of Gasteiger charges and cleanup of unwanted elements. The binding site was determined by the center of the specified residues in the pocket, and the searching space was set to 18.75 Å × 18.75 Å × 18.75 Å.
The overall VS workflow is depicted in Fig. 1c. The Specs library with around 210,000 small molecules was firstly pretreated by the Pan-Assay Interference Compounds (PAINS) rule [32] and Rapid Elimination Of Swill (REOS) rule [33] in Canvas to remove the noisy molecules with undesirable functional groups. Then, the Lipinski’s rule-of-five (Ro5) [34] and Opera rules [35] were used to eliminate non-drug-like molecules with the number of chiral centers ≥3, the violation count of the Oprea rule ≥3 and the violation count of Ro5 ≥2. Next, each molecule was prepared by the LigPrep module in Schrödinger to assign its ionized states, tautomers, stereoisomers and low-energy conformation. The ionized states and tautomers at pH = 7.0 ± 2.0 were generated by using the Epik method [36], and the maximum number of the stereoisomers for each molecule was set to 4. The other parameters for Ligprep were set to the default settings.
Because the binding site on the DBD is relatively flat, it is quite possible that the binding poses of molecules cannot be reliably predicted by a single docking program. In order to improve the prediction accuracy of binding poses, three docking programs (i.e., Glide SP, GOLD and AutoDock Vina) were used to conduct three individual docking calculations. Then, the pairwise root-mean-square-deviations (RMSDs) of the binding poses predicted by the three different programs for each molecule were calculated by the obrms utility in OpenBabel [37]. If two of the three RMSD values for a molecule were less than 2.0 Å, this molecule would be considered in further analysis, and otherwise abandoned. The above operations might lead to the loss of some potential binders, but to some extent they could enhance the reliability of VS. Next, the remaining molecules were ranked by integrating the three docking scores, and the 171 compounds with the docking scores higher than the predefined cutoffs (−3.5, 55 and −6 for Glide SP, Gold CHEMPLP and Autodock Vina, respectively) were clustered based on the Tanimoto coefficients using the MACCS fingerprints. Finally, 61 molecules were selected through visual inspection for bioassays.
Materials
Compounds
VPC-14228 (4-(4-phenylthiazol-2-yl) morpholine) and the tested compounds were purchased from Specs (Maryland, USA), and enzalutamide was purchased from MedChemExpress (New Jersey, USA).
Cells and plasmids
All the cells were purchased from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). Human PCa cells LNCaP, 22RV1, DU145 and PC3 were cultured in RPMI-1640 and F-12K medium with 10% FBS, 100 IU/mL penicillin, and 100 μg/mL streptomycin, respectively. A549, 3T3, MCF-7 and HeLa cells were cultured in DMEM medium with 10% FBS, 100 IU/mL penicillin, and 100 μg/mL streptomycin. All the cells were cultured with 5% CO2 and 95% atmosphere in cell culture incubator at 37 °C.
pCMV-GR11 (Addgene, #89105), pcDNA3-PRB (Addgene, #89130) and pCMV-hERalpha (Addgene, #101141) were gifts from Elizabeth Wilson. 3×ERRE/ERE-luciferase (Addgene, #37852) was a gift from Rebecca Riggins. GRE promoter was cloned into the BmtI and BglII sites of pNF-κB-luc (Beyotime Biotechnology, Cat #D2206, China). ARR3tk promoter was cloned into the HindIII and XhoI sites of PGL4.18 vector (Promega, USA).
Protein expression and purification
The AR DBD (residues: 561-679) was cloned into the NdeI and XhoI sites of pET28ɑ vector. The sequence of the AR DBD is HMCLICGDEASGCHYGALTCGSCKVFFKRAAEGKQKYLCASRNDCTIDKFRRKNCPSCRLRKCYEAGMTLGARKLKKLGNLKLQEEGEASSTTSPTEETTQKLTVSHIEGYECQPIFLNVLE. The plasmid with the mutated residue (Tyr594Asp) in the predicted binding site was constructed with the PCR technology. The forward cloning primer was 5ʹ-CAGAAGGACCTGTGCGCCAGCAGAAATGATTGC-3ʹ, and the reverse cloning primer was 5ʹ-GCACAGGTCCTTCTGTTTCCCTTCAGCGGCTCT-3ʹ (the mutated residues are bold). The plasmids were transformed into E. coli BL21 (DE3).
Cultures were grown to an OD600 of 0.8 in two liters of LB media supplemented with 50 μg/mL Kanamycin before induced with 0.1 mM IPTG for 3 h at 37 °C. The cells were collected by centrifugation at 4000 r/min for 15 min and then resuspended in 20 mL buffer A (50 mM Tris-HCl, pH=8.0, 300 mM NaCl, 5% glycerol) supplemented with 10 mM imidazole and 0.1% PMSF. Cell lysis was achieved by sonication, followed by centrifugation at 14500 × g for 30 min at 4°C. The protein was purified with HisPur™ Ni-NTA Resin (Thermo Scientific™; USA) and dialyzed with buffer A.
Cell culture, transfection, and luciferase reporter assays
In luciferase reporter assays, cells were cultured in 5% CSS RPMI-1640 (PC3) or DMEM media (A549 or HeLa) with 1 × 104 cells/well in 96-well plates for 24 h respectively. PC3 cells were transfected with 50 ng of hPR, 50 ng of ARR3tk-luciferase and 5 ng Rencilla by lip3000 transfection reagent (Thermo Scientific™, Cat #L3000075) for 24 h. The transfection procedure was also applied for the GR and ER assays, meanwhile HeLa and MCF-7 cells were transfected with 71 ng of GRɑ, 24 ng of MMTV-luciferase or 71 ng of ERɑ, 24 ng of ERRE/ERE-luciferase respectively. The bioactivity of GR, PR or ER was then stimulated with 1 nM dexamethasone, 10 nM progesterone and 1 nM estradiol, respectively. Cells were harvested by addition of 1× Passive Lysis Buffer (Promega, Cat # E1910), and luciferase activity was assayed by the Dual-Glo Luciferase system (Promega, Cat # E1910). Data were plotted as firefly luciferase activity normalized to Renilla luciferase activity in Relative Luciferase Units. Control wells with DMSO or steroid were included on each plate to define the 0% and 100% effect, respectively. Raw data values were transformed to % activation using the following equation:
AR transcriptional activity assay
A cell line of LNCaP that stably expresses eGFP under the regulation of an androgen response element was generated to investigate the agonist/antagonist activity of the selected compounds as previously described [38, 39]. The LNCaP-ARR2PB-eGFP cells were grown in phenol-red-free RPMI-1640 supplemented with 5% charcoal stripped serum (CSS) for 5 days. Then cells were seeded into a 96-well plate (3.5 × 104 cells per well) for 24 h, and then treated by a single concentration or increasing concentrations (0–50 μM) of compounds with 10 nM DHT for screening. The fluorescence was measured after incubating for 72 h (Synergy H1, BioTek. Excitation, 485 nm; Emission, 535 nm).
Prostate specific antigen (PSA) assay
The PSA secreted into the media was evaluated in parallel with the AR transcriptional activity assay using the same samples. After incubation of 3 days, 400 μL of the media was collected and sent to Cancer Hospital of University of Chinese Academy of Sciences, Zhejiang Cancer Hospital (Hangzhou, Zhejiang). The PSA level was evaluated with the IMMULITE®2000 XPi immunoassay system (Siemens Ltd, Erlangen, Germany).
Cell proliferation assay
Cell proliferation was measured using the 3-(4,5-dimethythiazol-2-yl)−2,5-diphenyl-2-H-tetrazolium bromide (MTT) colorimetric assay. A total of four types of PCa cell lines were involved, including LNCaP, 22RV1, PC3 and DU145. These cells were cultured in RPMI-1640 media (5% CSS) with 2×103-5 × 103 cells/well in 96-well plates (5 × 103 for LNCaP, 3 × 103 for 22RV1, and 2 × 103 for PC3 and DU145). After incubation at 37 °C for 24 h, cells were treated with serial dilutions of the tested compounds (10 nM DHT in extra for LNCaP culture) and incubated for another 3 days. Afterward, 10 μL of 5 mg/mL MTT solution was added into each well and incubated for another 4 h. Then, 100 μL of triplex 10% SDS-5% isobutyl alcohol-0.012 mol/L HCl (w/v/v) solution was added to dissolve the formazan crystals. The absorbance at 570 nm was measured with the reference wavelength at 650 nm using a spectrophotometer (Bioteck Eon, Winooski, VT).
Cell cytotoxicity assay
To rule out the inherent toxicity of the tested compounds, A549 and 3T3 cell lines were seeded in DMEM media at a density of 2× 103 cells per well and treated with the concentration gradient of the tested compounds for 24 h. Cell viability was then measured as described above.
Androgen displacement assay
Androgen displacement was assessed with the PolarScreenTM Androgen Receptor Competitor Green Assay Kit following the instructions of the manufacturer (Invitrogen Life Technologies, Inc.).
BLI assay
BLI experiments were conducted on a FortéBio Octet Red system. The biotinylated ARE was formed by annealing the following complementary oligonucleotides in H2O: upper strand, 5ʹ-Biotin-TACAAATAGGTTCTTGGAGTACTTTACTAGGCATGGACAATG-3ʹ, and lower strand, 5ʹ-CATTGTCCATGCCTAGTAAAGTACTCCAAGAACCTATTTGTA-3ʹ (AREs are bold). All experiments were conducted with the biotinylated ARE and purified AR DBD protein in PBST with 2.5% DMSO. The ARE (50 nM) was loaded onto streptavidin sensors in 200 μL PBS for 300 s, and then the ARE-loaded sensors were pre-equilibrated in PBS. The kinetics of ARE-protein association were monitored by soaking sensors into wells containing 1 μM AR DBD and 50 μM compound for 300 s, followed by dissociation in the same buffer deprived of protein for additional 180 s. All the experiments were carried out independently at least two times.
Confocal microscopy
LNCaP cells were seeded at a concentration of 5 × 104 per well on sterile coverslips placed within 12-well plates. After incubation at 37 °C for 24 h, cells were treated with 10 μM compounds for 12 h and then treated with 5 nM DHT for 1.5 h. After aspiration of the media, cells were fixed in 4% paraformaldehyde at 25°C for 20 min and incubated with AR antibodies (Cell Signaling Technology, Cat# 5153, Lot:1) overnight after washed with PBS three times. An Alexa-488 conjugated goat-anti rabbit lgG (Cell Signaling Technology, Cat# 4412, Lot: 20) diluted at 1:1000 was used as the secondary antibody. The counterstain DAPI was employed to visualize cell nucleus. Images were taken at 60 magnification using the Nikon A1R confocal spinning disk microscope and followed by the analysis with the NIS-Elements Viewer (Northern Eclipse, Empix Imaging, Inc.).
Western blotting
LNCaP cells were cultured in phenol-red-free RPMI-1640 (5% CSS) for 2 days and then seeded in 6-well plates (3 × 105 cells per well). After overnight incubation, the cells were treated with 10 μM of compounds under the presence of 10 nM DHT. After 48 h incubation, the cells were lysed with RIPA and subjected to 10% SDS-polyacrylamide for Western blotting. Protein was transferred to the methanol charged PVDF membrane and probed with rabbit anti-AR (Cell Signaling Technology, Cat# 5153, Lot:1) and mouse anti-TMPRSS2 (Santa Cruz Biotechnology, Cat# sc-101847, Lot: J1619J), along with monoclonal anti-GAPDH (Beyotime, Cat# AF1186, Lot: D525AA0002) utilized to show equal loading. Proteins were visualized using anti-mouse (Sangon Biotech, Cat# D110087, Lot: F910AA0025) or anti-rabbit HRP-conjugated secondary antibodies (Sangon Biotech, Cat# D110058-0100, Lot: F902AA0024). The protein bands were detected using the Automatic Gel Imaging Analysis System developer (Peiqing Science and Technology, Guangzhou, China).
Quantitative real-time reverse transcription polymerase chain reaction (Q-PCR)
Total RNA was isolated from cells using the EZ-10 DNAaway RNA Mini-Preps Kit (Sangon Biotech, Shanghai, China) according to the instructions of manufacturer. cDNA was generated using the Hifair® III 1st Strand cDNA Synthesis SuperMix (YEASEN, Shanghai, China). Diluted cDNA was mixed with the forward primer, reverse primer, SYBR Green PCR Master Mix (YEASEN, Shanghai, China), and RNase-free water in a 96-well plate. Analysis of mRNA expression was carried out using the Applied Biosystems QuantStudio 3. All samples were normalized to the level of GAPDH. The threshold cycles (Ct) for the control (GAPDH) and gene of interest were determined, and the relative mRNA levels were calculated by the 2−△△Ct method. The details of the primer sequences used in the study are shown in the supporting information.
RNA-seq
DU145 cells were cultured in RPMI-1640 media and then exposed to DMSO and 10 μM Cpd39, respectively, for 48 h. Total RNA was extracted with Trizol Reagent and sequenced using the Hiseq-PE150 high-throughput sequencing platform (Novogene, Beijing, China). The raw expression data were processed and normalized as followed.
First, the human reference genome GRCh37 (version 19) was downloaded from the Ensembl website (https://www.ensembl.org), a database project providing access to human genome annotation [40]. Four FASTQ files containing the paired-end sequence reads were aligned to the human reference genome using HISAT2 (Hierarchical Indexing for Spliced Alignment of Transcripts) (version 2.1.0) [41]. The output SAM (sequencing alignment/map) files were converted to the BAM (binary alignment/map) files and sorted using SAMtools (version 1.9) [42]. Next, StringTie [43] (version 2.0) was used to assemble the genes for the data set in each BAM file separately, calculate the expression levels of each gene and each isoform, and then merge all the gene structures found in any of the samples together using the full set of assemblies. The merged transcripts were passed to StringTie again so that it can re-calculate the transcript abundances. Finally, the Ballgown package [44] grouped all the transcripts and abundances from StringTie by experimental condition, and determined the expressed genes (DEGs) between conditions using a linear model for a log transformation of the FPKM values attached to transcripts. DEGs were defined as those with the false discovery rate (FDR) adjusted P-value less than 0.05 and the fold change between conditions more than 2. To identify the biological processes and pathways that were significantly enriched by DEGs, the gene list was analyzed using the Database for Annotation, Visualization and Integrated Discovery (DAVID) (version 6.8) [45] and KOBAS 2.0 [46].
Data and statistical analysis
Statistical analyses were performed with the GraphPad Prism software v 6.0 (San Diego, CA, USA). The differences of two groups were analyzed using a two-tailed Student’s t test, and the differences between more than two groups were analyzed using one-way analysis of variance (ANOVA), followed by Dunnett’s post hoc test. Some results were normalized to the control to avoid unwanted sources of variation. All data were presented as means ± SEM. *P < 0.05 was considered to be significant. The capacitive transients of some traces in the figures were trimmed for clarity.
Results and discussion
Evaluation of candidate compounds
The binding pocket formed by Ser579, Val582, Phe583, Arg586, Arg609, Lys610, Pro613 and Arg616 on the DBD-ARE binding interface was detected as the potential binding site, and then the Specs chemical library was virtually screened (Fig. 1c). The 61 compounds identified from the VS were tested for their antagonistic activities using a nondestructive enhanced green fluorescent protein (eGFP) assay [38]. Vpc-14228, a reported compound targeting the AR DBD, and enzalutamide were used as positive controls [22]. At first, the 61 compounds were tested at the concentration of 5 µg/mL, and compound Cpd31, Cpd34, Cpd55 and Cpd39, showed over 50% antagonistic activity of enzalutamide. Further testing under a concentration gradient (0.1-100 µM) illustrated that Cpd31 (IC50 = 39.46 µM) and Cpd39 (IC50 = 10.94 µM) exhibited a dose-dependent inhibition (Fig. 1d). Since Cpd39 showed better activity than Cpd31 (Fig. S1), it was selected as a potential antagonist hit for further studies. To avoid false-positive detection by the eGFP assay, a complementary PSA assay was employed [22]. PSA, an AR downstream target gene of AR, is a clinically important serum biomarker for PCa and has been widely used as an indicator to evaluate AR transcriptional activity. And Cpd39 inhibited the expression of PSA in a dose-dependent manner with IC50 = 20.16 µM and those for Vpc-14228 and enzalutamide were 1.26 µM and 0.329 µM, respectively (Fig. 1d and e).
Structure activity relationship (SAR) analysis for the analogs of Cpd39
To search for more potent analogs and explore their initial SAR of the new scaffold of Cpd39, similarity- and substructure-based analog searching was performed using Cpd39 as the query. A total 27 analogs of Cpd39 were then identified in the ChemDiv library for activity assessment. The chemical structures of these molecules and their bioactivities are summarized in Table S1. In general, no significant activity improvement was observed, and only Cpd39-12 showed comparable bioactivity to Cpd39. However, valuable information can be captured from the preliminary SAR analysis, which would provide helpful guidance for further structural optimization of Cpd39. According to the predicted binding mode of Cpd39 within the binding pocket on the DBD (Fig. 1f and g), the carboxyl group on the phenyl ring could form two H-bonds with Arg586 and a H-bond with Ser579, and the amide group on the linker could form a H-bond with Arg616. These H-bond interactions are most likely the primary driving force for the binding of Cpd39. Apart from the H-bond interactions, the hydrophobic interactions between Cpd39 and some surrounding residues should also contribute a lot. For example, the phenyl ring linked to the carboxyl group is located exactly in the center of Phe583, Tyr594, and Pro613, and thereby can form favorable van der Waals interactions with the hydrophobic side chains of these residues. In addition, the alkyl group in another phenyl ring may also play an important role in the binding by forming favorable hydrophobic interactions with Lys606 and Phe607. Another important factor may lie on the existing of Arg586 and Lys610, which can construct a natural groove on the surface to fit the binding of Cpd39. The bioactivities of the analogs are well consistent with the binding mode analysis. The ortho substituents of H-bond acceptors on Ring A, such as the carboxylic group, ester group and amide group, were favorable for ligand binding as the validation of the binding mode. For the analogs without any ortho substituent on Ring A, including Cpd39-1, Cpd39-2, Cpd39-3, Cpd39-10, Cpd39-11, Cpd39-20, and Cpd39-23, were almost inactive due to the absence of the crucial H-bond interactions with Arg586 or Ser579. The size and flexibility of the substituents on Ring C are also critical to ligand binding. Larger substituents, such as tert-pentyl (Cpd39) and tert-butyl (Cpd39-9 and Cpd39-12), led to enhanced binding affinity, while smaller substituents, such as isopropyl (Cpd39-4 and Cpd39-5), methyl (Cpd39-6) and halogen atoms (Cpd39-17 and Cpd39-25), did not show favorable effect. As for the compounds Cpd39-8, Cpd39-16 and Cpd39-24, their poor activities most likely stemmed from the rigidness of their Ring C. The 2D presentation (Fig. 1h) showed that only the substituent groups with appropriate size and flexibility can stretch into the groove formed by Arg586 and Lys610. Regarding the other analogs, the reason of their low activities needs to be further explored.
Cpd39 effectively inhibits AR-mediated PCa cell proliferation
To figure out the antitumor effect of Cpd39 on AR-expressing PCa cells, LNCaP, an androgen-dependent cell line, and 22RV1, an androgen-insensitive cell line, were utilized for testing [47]. 22RV1 cell line is derived from the CWR22 xenograft, which expresses high level of AR-V7 and is resistant to enzalutamide. As a result, Cpd39 exerted a dose-dependent manner to inhibit the growth of LNCaP and 22RV1 cells (Fig. 2a and b). At a lower concentration of 1 µM, the potency of Cpd39 was worse than those of the controls, but at higher concentrations, Cpd39 outperformed both Vpc-14228 and enzalutamide. Cpd39 could suppress the cell viability of LNCaP to 75% at 10 μM and even less than 20% at 50 μM. In addition, Cpd39 was more effective than Vpc-14228 and enzalutamide in suppressing 22RV1 growth. Upon the treatment of 20 μM and 50 μM Cpd39 for 72 h, the growth of 22RV1 cells was significantly suppressed to 85% and 50%, respectively. In contrast, Vpc-14228 exhibited a minor effect on the growth of 22RV1.

a, b The antiproliferative effect of Cpd39 on LNCaP and 22RV1 cells using MTT assay (n = 4). c–f The antiproliferative effects of Cpd39 on four cell lines, including PC3, DU145, 3T3, and A549 cells (n = 3). g Distribution of the DU145 transcriptome sequences among KEGG pathways (n = 2). h The mRNA expression levels of RHOA, PI3KCB, ERBB2, METTL3, and EIF4B in DU145 cells (n = 3). *P < 0.05 versus DMSO group (n = 5 in each group).
Cpd39 inhibits the proliferation of AR-independent PCa cell with no toxicity
To determine whether the anti-proliferative activity of Cpd39 is caused by AR down-regulation, the compound was further evaluated on two androgen-independent cell lines, DU145 and PC3 (Fig. 2c and d). The cell viability curves of PC3 for Cpd39, Vpc-14228 and enzalutamide at different concentrations ranging from 0 to 50 µM have a similar shape. The IC50 values of enzalutamide and Vpc-14228 were higher than 100 µM for both PC3 and DU145, suggesting that the cytotoxicity of the compounds was specifically mediated through AR. However, Cpd39 could reduce cell viability to 50%–60% for PC3 cell line at 50 µM and the corresponding IC50 value was 47.98 µM. Cpd39 exhibited an even stronger inhibitory effect on the proliferation of DU145 (IC50 = 13.28 µM) than on PC3 cells. To assess whether the compound is inherently toxic, 3T3 mouse fibroblasts and A549 human lung adenocarcinoma cell lines were selected for in vitro cytotoxicity assessment. To our surprise, 50 μM of Cpd39 showed a negligible effect on both 3T3 and A549 cell lines. The IC50 values of Cpd39 for 3T3 and A549 cell lines were larger than 80 µM and 100 µM, respectively, suggesting that the toxicity of Cpd39 to PCa cells is specific (Fig. 2e and f). In addition, it is obvious that the antiproliferative effect of Cpd39 on AR negative cells PC3 and DU145 was not mediated through AR, which might be linked with some unknown mechanisms.
To explore the inhibition mechanism of Cpd39 toward DU145 cell line, HiSeq-PE150 was used to conduct sequencing analysis. The gene expression differences were analyzed by comparing the transcriptomes before and after the treatment of 10 μM Cpd39. At first, those up-regulated or down-regulated genes were examined to confirm whether Cpd39 could yield any potential genotoxic effect. Generally, the genes with genotoxicity including a number of p53 target genes and those involved in apoptosis, DNA repair, DNA damage response or stress response were checked. None of them showed significant response, indicating that Cpd39 is inherently nontoxic [48].
Enrichment analysis of Kyoto encyclopedia of genes and genomes (KEGG) pathways was also conducted to identify the essential pathways potentially involved. The results revealed that the “metabolic pathways”, “pathways in cancer”, “proteoglycans in cancer” and “ribosome” (Fig. 2g) were mainly affected by the treatment of Cpd39, suggesting that the changes in these pathways/processes might account for its anticancer efficacy. After an in-depth analysis, it was noticed that the mTOR and PI3K-AKT signaling pathways showed the most prominent differences, which might be the main reason why Cpd39 could induce the apoptosis of DU145 (Figs. S2 and S3). The genes encoding RHOA (Ras homolog family member A, fold change = 27.4, P = 0.013), PI3KCB (phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit beta, fold change = 470.1, P = 0.044) and ERBB2 (erb-b2 receptor tyrosine kinase 2, fold change = 44.8, P = 0.012) were synergistically trans-repressed. All of them are involved in the PI3K and AKT signaling networks, which were known to contribute significantly to the CRPC development [49]. In fact, combing AR antagonists with numerous inhibitors of the PI3K-AKT-mTOR pathway has shown activity in preclinical models, and related clinical trials are under study in CRPC [50,51,52,53]. RhoA, a member of the Rho family of GTP-binding proteins, is over-expressed in many forms of malignant cancers and plays an important role in focal adhesion regulation, actin stress fiber formation, and cell migration of castration-resistance transformation of PCa [54]. PI3KCB is the gene that encodes the p110β lipid kinase catalytic subunit of PI3Kβ. PI3Kβ is a critically important factor in the PI3K signaling pathway and can drive tumorigenic cell growth and migration in several tumor types. To demonstrate the efficacy of Cpd39, Q-PCR was performed to validate whether these three key genes were truly trans-repressed. The results revealed Cpd39 could significantly down-regulate the expression of these three genes and could exert a deep effect on the growth and metastasis of malignant cells, with no cytotoxicity induced (Fig. 2h). Taken together, we deduced that the antigrowth effect of Cpd39 on DU145 probably owes to its influence on the PI3K and AKT signaling pathways.
Cpd39 is a novel AR antagonist targeting AR DBD
To validate whether the antitumor potency of Cpd39 is attributed to the disruption of the binding of the AR DBD to ARE, BLI, a label-free technology was utilized. BLI has been widely employed in drug discovery and life science studies in vitro to measure biomolecular interactions. In this study, we initially attempted to confirm whether the compound could affect the binding of the AR DBD to dsDNA bearing the ARE2 sequence at a concentration of 50 µM, with enzalutamide and Vpc-14228 as the references. In this task, the biotinylated dsDNA (ARE) was attached to the streptavidin BLI sensors, and then exposed to the solution containing both of the AR DBD and tested compound, which is capable of resulting in a large shift for the signal of wavelength compared to the solution containing only the AR DBD if the compound does interrupt the interaction between the AR DBD and ARE. And the behaviors of Cpd39 and Vpc-14228 were similar and the signal decreased ~12%–15% in comparison with the AR DBD alone (Fig. 3a–c). In contrast, the wavelength change generated by enzalutamide was little, which supported the fact that enzalutamide had no ability to impede the binding between the AR DBD and ARE. To further understand the inhibition behavior of Cpd39, this experiment was carried out by testing a set of concentrations of the compound. The inhibitory activity of Cpd39 steadily increased with the increase of concentrations, and Cpd39 could even impede the interaction between the AR DBD and ARE at a concentration of 500 µM (Fig. 3d). The behavior of Cpd39 was dissimilar to that of Vpc-14228 (Fig. 3e), as Vpc-14228 functioned back at 500 µM. Furthermore, to confirm the location of the binding site, we performed the same BLI experiments with the mutated AR DBD protein. Previous studies showed that the Tyr594Asp mutation at the DBD-ARE binding interface could maintain the AR transcriptional activity [22], so this Tyr594Asp mutated AR DBD was used in this study. In our experiments, both of Cpd39 and Vpc-14228 lost their ability to block the binding of the mutated AR DBD to ARE. The association kinetic result of dsDNA with the mutated AR DBD was significantly different from that of dsDNA with the wild type AR DBD (Fig. 3f and g). That is to say, similar to Vpc-14228, Cpd39 was unable to affect either the association or dissociation of dsDNA with the mutated protein, indicating that Cpd39 did target on the site of Tyr594 at the DBD-ARE binding interface. Together, Cpd39 could inhibit the protein-DNA interactions in vitro and bind to the proposed site at the AR DBD.

a–c Biotinylated ARE was loaded onto streptavidin sensors for bilayer interferometry analysis. t = 0 s. BLI sensors preloaded with ARE (50 nM) are baselined in buffer with the indicated compound (50 μM); at the first inflection point (arrow), Sensors are moved to a solution of the AR DBD (1 μM) alone (2.5% DMSO), or the AR DBD mixed with tested compounds for 300 s, and then moved back to buffer for dissociation. The y axis represents the nanometer shift in wavelength resulting from ligand (dsDNA) binding/dissociation (n = 2). d, e Same as (b, c), but with different concentrations of tested compounds (0–500 μM). The BLI results reflected the direct binding of the compounds to the AR DBD protein (n = 2). f–g Same as (b, c), but with the Y594D mutated AR DBD (n = 2). Non-specific interactions between the protein or compounds and sensors were subtracted from the BLI signal.
In addition, we also ruled out the possibility of the binding of these compounds to the AR DBD or ARE individually (Fig. S4). The Kd value between the AR DBD and Cpd39 was 6.3 × 10-3 M, which was usually regarded as a weak interaction (Fig. S4a and S4b). And Cpd39 did not show a direct interaction with ARE (Fig. S4c).
The effects of Cpd39 on other steroid hormone receptors
AR together with GR, ER, and PR belongs to the steroid hormone receptor subfamily of the nuclear receptor super family. They recognize a similar DNA response element to play the role of transcriptional factors. To determine whether Cpd39 cross-reacts with the DBDs of the related nuclear receptors, we performed luciferase assays with full-length GRɑ, ERɑ, and PR. Luciferase constructs contained the corresponding response regions of glucocorticoid-response element for GR, a hybrid ERRE/ERE element for ER, and ARR3tk for PR. Cpd39 exhibited inhibitory activity on GR, no significant effects on ERɑ, and agonist activity on PR, while Vpc-14228 stimulated GR, ER, and PR in varying degrees (Fig. 4a and b). In contrast, enzalutamide showed almost no effect on GR, weak inhibition on ER at high concentration, and somewhat inhibitory activity on PR (Fig. 4c). In our laboratory, the selectivity of enzalutamide demonstrated a same tendency as previous report, while Vpc-14228 showed somehow differently [55]. Such discrepancy may be explained by several factors. This includes the dissimilarities of the cell lines, the reporter plasmids and the experimental schemes. Indeed, selectivity is a challenge for the drugs targeting the steroid hormone receptors. Even marketed drugs such as enzalutamide cannot exclude cross activity with the other unexpected steroid hormone receptors. Nevertheless, considering the diversified expression levels of the steroid hormone receptor during PCa progression, for example, an antagonist targeting both AR and GR may treat PCa at late stage [4], clarifying the specificity of a certain compound is more important than searching for a highly specific one at the initial stage of a drug development. Here, in our selectivity assays, simply compare specificity, enzalutamide performed the best, and Cpd39 displayed relatively better than Vpc-14228.

a–c Cross-reactivity analysis of Cpd39 inhibition. d Effect of compounds on AR nuclear localization. Microscopic images of LNCaP cells treated with 10 μM compounds and 5 nM DHT or DMSO only (magnification ×600; scale bar = 20 μm) (n = 3). e The mRNA expression levels of AR target genes including KLK3 (PSA), TMPRSS2, ACPP, and FKBP5 analyzed by quantitative analysis after LNCaP cells treated with 10 µM compounds and 10 nM DHT. *P < 0.05 versus DHT group (n = 3 in each group). f Protein expression of AR and TMPRSS2 in LNCaP analyzed by Western blot analysis (n = 2). g Expression of AKT1 analyzed by quantitative analysis after 22RV1 cells were treated with indicated compounds. *P < 0.05 versus DMSO group (n = 5 in each group).
Cpd39 does not target LBD or block AR nuclear localization
We then attempted to exclude the possibility that Cpd39 binds to the traditional LBP site. Accordingly, a PolarScreenTM AR competitor assay was carried out. This assay was conducted for Cpd31 and Cpd39 at a concentration of 10 μM with DMSO and DHT as the controls. The behavior of Cpd39 was similar to that of the vehicle control (DMSO) but significantly different from that of the positive control (DHT), supporting that Cpd39 does not interact with the LBP (Fig. S4d). In addition, the translocation of AR into nucleus is a key step in AR signaling. It has been proven that some of the drugs targeting the AR LBD are able to exert their functions by blocking the nuclear translocation of AR, thus preventing the acceptor from the initiation of transcription [55,56,57]. However, the compounds targeting the DBD should theoretically function in the nucleus to block the recognition of AR to ARE. Therefore, we utilized the confocal assay to evaluate whether Cpd39 could affect the DHT-induced translocation of AR from the cytoplasm to the nucleus. The group without the treatment of DHT or enzalutamide showed low levels of AR in the nucleus compared with the group only treated with DMSO (Fig. 4d). Conversely, VPC-14228 and Cpd39 could not prevent the DHT-stimulated nuclear localization of AR with considerable fluorescence signal observed in the nucleus. The results indicated that Cpd39 could not impede the DHT-induced AR nuclear translocation, supporting for the direct action of Cpd39 on the DBD.
Cpd39 down regulates not only the AR but also ARVs responsive genes in PCa Cells
To assess the ability of blocking the transcription of AR regulated genes, Cpd39 was tested on LNCaP cells. The mRNA levels of three AR target-genes [58, 59] KLK3 (PSA), TMPRSS2 and FKBP5 were evaluated by Q-PCR. In our study, KLK3, TMPRSS2 and FKBP5 are androgen-upregulated genes and their mRNA levels substantially decreased compared to that of the DHT control (Fig. 4e). Somehow, the gene of ACPP previously reported as an androgen-downregulated gene did not show expected higher transcriptional level when treated with Cpd39. To further confirm the antiandrogenic activity of Cpd39, the Western blot analysis was carried out to check its effect on the expression of TMPRSS2 protein and AR (Fig. 4f). Consistent with the result of Q-PCR, the expression of TMPRSS2 protein was down-regulated at the presence of Cpd39, while AR seems unaffected.
Furthermore, to investigate whether Cpd39 has an impact on AR-V7 dependent genes, ARVs over-expressed cell line of 22RV1 was selected for testing. The cells were treated with 10 μM and 50 μM Cpd39 in charcoal-stripped serum (5% CSS)-containing medium which represented an androgen-independent state. AKT1, reported preferentially as a subset of genes uniquely upregulated by ARVs rather than the prototype AR [60], was analyzed by Q-PCR. Remarkably, in our test AKT1 was downregulated by Cpd39 at both the concentrations of 10 μM and 50 μM compared to the control group (Fig. 4g), though the effect is significant only under 10 μM. And DHT has no effect. Based on the results above, we infer that Cpd39 is a novel AR antagonist capable of fighting ARVs.
Conclusion
In order to overcome the acquired resistances of approved AR antagonists, new treatments with non-conventional mechanisms are required. The binding of AR DBD to ARE is essential for the activation of both AR and ARVs. Targeting the interaction site, the P-box of AR DBD, between AR DBD with ARE is a promising strategy to overcome the drug resistances derived from gained mutations at the AR LBD. So far, only a chemical scaffold has been discovered to target the P-box of AR DBD and play the role of AR antagonist. Our study discovered a novel AR DBD-ARE blocker Cpd39 with new scaffold, providing a promising hit for the treatment of PCa. However, Cpd39 still needs extensive structural optimization to improve activity, and our studies on Cpd39 analogs have provided some referential information.
References
Huggins C, Stevens RE, Hodges CV. Studies on prostate cancer II the effects of castration on advanced carcinoma of the prostate gland. Arch Surg-Chic. 1941;43:209–23.
Bohl CE, Gao WQ, Miller DD, Bell CE, Dalton JT. Structural basis for antagonism and resistance of bicalutamide in prostate cancer. Proc Natl Acad Sci USA. 2005;102:6201–6.
Li YM, Chan SC, Brand LJ, Hwang TH, Silverstein KAT, Dehm SM. Androgen receptor splice variants mediate enzalutamide resistance in castration-resistant prostate cancer cell lines. Cancer Res. 2013;73:483–9.
Li D, Zhou WF, Pang JP, Tang Q, Zhong BL, Shen C, et al. A magic drug target: androgen receptor. Med Res Rev. 2019;39:1485–514.
Chen CD, Welsbie DS, Tran C, Baek SH, Chen R, Vessella R, et al. Molecular determinants of resistance to antiandrogen therapy. Nat Med. 2004;10:33–9.
Tan MHE, Li J, Xu HE, Melcher K, Yong EL. Androgen receptor: Structure, role in prostate cancer and drug discovery. Acta Pharmacol Sin. 2015;36:3–23.
Shafi AA, Yen AHE, Weigel NL. Androgen receptors in hormone-dependent and castration-resistant prostate cancer. Pharmacol Ther. 2013;140:223–38.
Ning YMM, Pierce W, Maher VE, Karuri S, Tang SH, Chiu HJ, et al. Enzalutamide for treatment of patients with metastatic castration-resistant prostate cancer who have previously received docetaxel: U.S. Food and drug administration drug approval summary. Clin Cancer Res. 2013;19:6067–73.
Chi KN, Agarwal N, Bjartell A, Chung BH, AJPD Gomes, Given R, et al. Apalutamide for metastatic, castration-sensitive prostate cancer. N Engl J Med. 2019;381:13–24.
Fizazi K, Shore N, Tammela TL, Ulys A, Vjaters E, Polyakov S, et al. Darolutamide in nonmetastatic, castration-resistant prostate cancer. N Engl J Med. 2019;380:1235–46.
Watson PA, Arora VK, Sawyers CL. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat Rev Cancer. 2015;15:701–11.
Miller K. Re: Antitumour activity of MDV3100 in castration-resistant prostate cancer: A phase 1-2 study. Eur Urol. 2010;58:464–5.
Joseph JD, Lu N, Qian J, Sensintaffar J, Shao G, Brigham D, et al. A clinically relevant androgen receptor mutation confers resistance to second-generation antiandrogens enzalutamide and ARN-509. Cancer Discov. 2013;3:1021–9.
Dehm SM, Schmidt LJ, Heemers HV, Vessella RL, Tindall DJ. Splicing of a novel androgen receptor exon generates a constitutively active androgen receptor that mediates prostate cancer therapy resistance. Cancer Res. 2008;68:5469–77.
Hornberg E, Ylitalo EB, Crnalic S, Antti H, Stattin P, Widmark A, et al. Expression of androgen receptor splice variants in prostate cancer bone metastases is associated with castration-resistance and short survival. PLoS One. 2011;6:e19059.
Caboni L, Kinsella GK, Blanco F, Fayne D, Jagoe WN, Carr M, et al. “True” antiandrogens-selective non-ligand-binding pocket disruptors of androgen receptor-coactivator interactions: novel tools for prostate cancer. J Med Chem. 2012;55:1635–44.
Ban FQ, Leblanc E, Li HF, Munuganti RSN, Frewin K, Rennie PS, et al. Discovery of 1H-indole-2-carboxamides as novel inhibitors of the androgen receptor binding function 3 (BF3). J Med Chem. 2014;57:6867–72.
Munuganti RSN, Hassona MDH, Leblanc E, Frewin K, Singh K, Ma D, et al. Identification of a potent antiandrogen that targets the BF3 site of the androgen receptor and inhibits enzalutamide-resistant prostate cancer. Chem Biol. 2014;21:1476–85.
Khorasanizadeh S, Rastinejad F. Nuclear-receptor interactions on DNA-response elements. Trends Biochem Sci. 2001;26:384–90.
Shaffer PL, Jivan A, Dollins DE, Claessens F, Gewirth DT. Structural basis of androgen receptor binding to selective androgen response elements. Proc Natl Acad Sci USA. 2004;101:4758–63.
Lim MY, Otto-Duessel M, He ML, Su L, Nguyen D, Chin E, et al. Ligand-independent and tissue-selective androgen receptor inhibition by pyrvinium. Acs Chem Biol. 2014;9:692–702.
Li HF, Ban FQ, Dalal K, Leblanc E, Frewin K, Ma D, et al. Discovery of small-molecule inhibitors selectively targeting the DNA-binding domain of the human androgen receptor. J Med Chem. 2014;57:6458–67.
Ban FQ, Dalal K, Li HF, LeBlanc E, Rennie PS, Cherkasov A. Best practices of computer-aided drug discovery: Lessons learned from the development of a preclinical candidate for prostate cancer with a new mechanism of action. J Chem Inf Model. 2017;57:1018–28.
Dalal K, Ban FQ, Li HF, Morin H, Roshan-Moniri M, Tam KJ, et al. Selectively targeting the dimerization interface of human androgen receptor with small-molecules to treat castration-resistant prostate cancer. Cancer Lett. 2018;437:35–43.
Sastry GM, Adzhigirey M, Day T, Annabhimoju R, Sherman W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J Comput Aided Mol Des. 2013;27:221–34.
Kaminski GA, Friesner RA, Tirado-Rives J, Jorgensen WL. Evaluation and reparametrization of the OPLS-AA force field for proteins via comparison with accurate quantum chemical calculations on peptides. J Phys Chem B. 2001;105:6474–87.
Banks JL, Beard HS, Cao YX, Cho AE, Damm W, Farid R, et al. Integrated modeling program, applied chemical theory (impact). J Comput Chem. 2005;26:1752–80.
Olsson MHM, Sondergaard CR, Rostkowski M, Jensen JH. Propka3: Consistent treatment of internal and surface residues in empirical pK(a) predictions. J Chem Theory Comput. 2011;7:525–37.
Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, et al. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem. 2004;47:1739–49.
Jones G, Willett P, Glen RC, Leach AR, Taylor R. Development and validation of a genetic algorithm for flexible docking. J Mol Biol. 1997;267:727–48.
Trott O, Olson AJ. Software news and update autodock vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31:455–61.
Baell JB, Holloway GA. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J Med Chem. 2010;53:2719–40.
Walters WP, Namchuk M. Designing screens: how to make your hits a hit. Nat Rev Drug Discov. 2003;2:259–66.
Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 1997;23:3–25.
Oprea TI. Property distribution of drug-related chemical databases. J Comput Aided Mol Des. 2000;14:251–64.
Shelley JC, Cholleti A, Frye LL, Greenwood JR, Timlin MR, Uchimaya M. Epik: a software program for pK (a) prediction and protonation state generation for drug-like molecules. J Comput Aided Mol Des. 2007;21:681–91.
O’Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR. Open babel: An open chemical toolbox. J Cheminformatics. 2011;3:1–12.
Tavassoli P, Snoek R, Ray M, Rao LG, Rennie PS. Rapid, non-destructive, cell-based screening assays for agents that modulate growth, death, and androgen receptor activation in prostate cancer cells. Prostate. 2007;67:416–26.
Zhou WF, Duan MJ, Fu WT, Pang JP, Tang Q, Sun HY, et al. Discovery of novel androgen receptor ligands by structure-based virtual screening and bioassays. Genom Proteom Bioinf. 2018;16:416–27.
Hubbard T, Barker D, Birney E, Cameron G, Chen Y, Clark L, et al. The ensembl genome database project. Nucleic Acids Res. 2002;30:38–41.
Kim D, Landmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 2015;12:357–60.
Khalil AM, Guttman M, Huarte M, Garber M, Raj A, Morales DR, et al. Many human large intergenic noncoding rnas associate with chromatin-modifying complexes and affect gene expression. Proc Natl Acad Sci USA. 2009;106:11667–72.
Pertea M, Pertea GM, Antonescu CM, Chang TC, Mendell JT, Salzberg SL. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol. 2015;33:290–5.
Frazee AC, Pertea G, Jaffe AE, Langmead B, Salzberg SL, Leek JT. Ballgown bridges the gap between transcriptome assembly and expression analysis. Nat Biotechnol. 2015;33:243–6.
Huang DW, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1–13.
Xie C, Mao XZ, Huang JJ, Ding Y, Wu JM, Dong S, et al. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011;39:W316–W22.
Sramkoski RM, Pretlow TG, Giaconia JM, Pretlow TP, Schwartz S, Sy MS, et al. A new human prostate carcinoma cell line, 22r upsilon 1. Vitr Cell Dev. 1999;35:403–9.
Ellinger-Ziegelbauer H, Fostel JM, Aruga C, Bauer D, Boitier E, Deng SB, et al. Characterization and interlaboratory comparison of a gene expression signature for differentiating genotoxic mechanisms. Toxicol Sci. 2009;110:341–52.
Li Q, Huang H, He Z, Sun Y, Tang YF, Shang XH, et al. Regulatory effects of antitumor agent matrine on FOXO and PI3K-AKT pathway in castration-resistant prostate cancer cells. Sci China Life Sci. 2018;61:550–8.
Carver BS, Chapinski C, Wongvipat J, Hieronymus H, Chen Y, Chandarlapaty S, et al. Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer Cell. 2011;19:575–86.
Bitting RL, Armstrong AJ. Targeting the PI3K/AKT/mTOR pathway in castration-resistant prostate cancer. Endocr Relat Cancer. 2013;20:R83–99.
Kato M, Banuelos CA, Imamura Y, Leung JK, Caley DP, Wang J, et al. Cotargeting androgen receptor splice variants and mTOR signaling pathway for the treatment of castration-resistant prostate cancer. Clin Cancer Res. 2016;22:2744–54.
Jamaspishvili T, Berman DM, Ross AE, Scher HI, De Marzo AM, Squire JA, et al. Clinical implications of PTEN loss in prostate cancer. Nat Rev Urol. 2018;15:222–34.
Thorne CA, Hanson AJ, Schneider J, Tahinci E, Orton D, Cselenyi CS, et al. Small-molecule inhibition of Wnt signaling through activation of casein kinase 1 alpha. Nat Chem Biol. 2010;6:829–36.
Dalal K, Roshan-Moniri M, Sharma A, Li HF, Ban FQ, Hessein M, et al. Selectively targeting the DNA-binding domain of the androgen receptor as a prospective therapy for prostate cancer. J Biol Chem. 2014;289:26417–29.
Ferraldeschi R, Pezaro C, Karavasilis V, de Bono J. Abiraterone and novel antiandrogens: overcoming castration resistance in prostate cancer. Annu Rev Med. 2013;64:1–13.
Rathkopf D, Scher HI. Androgen receptor antagonists in castration-resistant prostate cancer. Cancer J. 2013;19:43–9.
Nelson PS, Clegg N, Arnold H, Ferguson C, Bonham M, White J, et al. The program of androgen-responsive genes in neoplastic prostate epithelium. Proc Natl Acad Sci USA. 2002;99:11890–5.
Magee JA, Chang LW, Stormo GD, Milbrandt J. Direct, androgen receptor-mediated regulation of the FKBP5 gene via a distal enhancer element. Endocrinology. 2006;147:590–8.
Guo ZY, Yang X, Sun F, Jiang RC, Linn DE, Chen HG, et al. A novel androgen receptor splice variant is up-regulated during prostate cancer progression and promotes androgen depletion-resistant growth. Cancer Res. 2009;69:2305–13.
Acknowledgements
This study was supported by the Key R&D Program of Zhejiang Province (2020C03010), National Natural Science Foundation of China (81773632), Zhejiang Provincial Natural Science Foundation of China (LZ19H300001), and the Key R&D Program of Zhejiang Province (2019C02024).
Author information
Authors and Affiliations
Contributions
TJH and DL initiated and supervised the research. CS and JPP designed the experiments. CS, JPP, conducted virtual screening, compound validations and biological assays. WFZ, YXW, LHS, XC, YS, XPH, FZ, LX, DYZ, LX, and XHX helped performing part of biological assays. JPP, CS, DL, and TJH wrote the manuscript, and the other authors contributed specific parts of the manuscript. TJH and DL assume responsibility for the manuscript in its entirety. All authors have critically revised the manuscript and approved its final version.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Supplementary information
Rights and permissions
About this article

Cite this article
Pang, Jp., Shen, C., Zhou, Wf. et al. Discovery of novel antagonists targeting the DNA binding domain of androgen receptor by integrated docking-based virtual screening and bioassays. Acta Pharmacol Sin 43, 229–239 (2022). https://doi.org/10.1038/s41401-021-00632-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-021-00632-5
Keywords
This article is cited by
-
Discovery of a novel nonsteroidal selective glucocorticoid receptor modulator by virtual screening and bioassays
Acta Pharmacologica Sinica (2022)
